
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Research ArticleDOI Number : 10.36811/ijgmgt.2019.110004Article Views : 15Article Downloads : 17
Deletion of SHANK3 and CREBBP gene in the patients with intellectual disability and mix phenotype
Jamileh Malbin
ANAS Genetic Resources Institute, Baku, Azerbaijan, Genetic Counseling Center, State Welfare Organization, Uremia, Iran
Corresponding Author: Jamileh Malbin, Azerbaijan National Academy of Science (ANAS), Genetic Resources Institute, Baku, Azerbaijan; genetic counseling center, Urmia welfare organization of west Azerbaijan Iran; Tel 00989144418710; Email: jamaalbin@yahoo.com
Article Information
Aritcle Type: Research Article
Citation: Jamileh Malbin. 2019. Deletion of SHANK3 and CREBBP gene in the patients with intellectual disability and mix phenotype. Int J Genet Med Gene Ther. 1: 15-20.
Copyright: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2019; Jamileh Malbin
Publication history:
Received date: 13 August, 2019Accepted date: 27 August, 2019
Published date: 30 August, 2019
Abstract
Rubinstein-Taybi Syndrome (RSTS) as a group of congenital anomalies mainly include, short broad thumbs and toes, short stature and intellectual disability are caused by either a micro-deletion in the CREBBP (CBP) or EP300 genes. Generally most RSTS patients have a deletion in the CREBBP gene but some patients have shown deletion in the EP300 gene. Here we introduce an affected case without some typical characteristics of RSTS with deletions in the CREBBP and SHANK3 genes. The patient was a 24 years old man with a history of infantile hypotonia and childhood developmental delay, heavy eyebrows, ptosis, speech difficulty without large thumb and toes. The conventional cytogenetic finding was normal male. Further investigation was performed using Multiplex Ligation Probe Amplification (MLPA) technique to screen micro-deletion syndromes and subtelomeric rearrangements and Micro-deletion was detected in CREBBP and SHANK3 gene and a detected in DECR2 gene. Deletion in the CREBBP or EP300 genes or both in the patients with broad thumb and toes (RSTS) has been detected but there are other patients with deletion in CREBBP gene without this sign of RSTS. However, we report SHANK3 gene deletion in the patient with deletion in CREBBP gene and without broad thumbs and toes.
Keywords: Rubinstein-Taybi Syndrome (RSTS); CREBBP; SHANK3; Broad thumb and toes
Introduction
Rubinstein-Taybi Syndrome (RSTS) (OMIM: 180849) is a rare congenital anomaly with a frequency of approximately 1 in 100,000 to 125,000 newborns [1]. The most frequent symptoms in the people with typical RSTS include growth deficiency, delayed bone age, intellectual disability, abnormally broad thumbs and toes, breathing difficulties [2]. Some patients experience swallowing problems [3]. Another category of RSTS known as “atypical RSTS” (OMIM#180849) or RSTS2 includes patients with a multiple congenital anomalies such as intellectual disability, microcephaly, broad thumbs and halluces, and dysmorphic facial features [4]. Some symptom such as broad thumb and toesm ptosis, intellectual disability, developmental delay and speech difficulties seems to be remarkable in both types of RSTS but some other such as form of nose and dolichocephaly is different. More than 50% patients with RSTS has deletion at CREBBP gene and has been known as RSTS1 [5] and less than 3% of patients with RSTS has a deletion in EP300 gene and has been known as RSTS2 [6]. Although this syndrome considers as an autosomal dominant, most cases of RSTS reported as a sporadic and the families with two or more affected children is very rare [4,7].
The CREBBP gene with 31 exons, approximately 155 kb of genomic DNA is located on chromosome 16p13.3 and EP300 gene also contains 31 exons, located on chromosome 22q13.2. Both CREBBP and EP300 is transcriptional co-activator that involved in various signal transduction pathways [8,9]. Mutation in these two genes has been known as RSTS but in some cases RSTS symptom especially large thumb and toes has not noticed in the patients with mutation either in CREBBP [10] nor EP300 gene [11].
Clinical Report
The patient was a 26-years-old man with a history of infantile hypotonia and childhood developmental delay. He is last of 10 children of non- consanguineous parents; pedigree of this family has been shown in figure1. He has a history of epilepsy starting from 5th year of his life. His dysmorphic feature and clinical signs includes heavy eyebrows, ptosis, speech difficulty, high arched palate, high set ears spastic paraplegia in the upper body and narrow opening mouth; he has a surgery because of adhesion tongue still difficulties in feeding (figure2a). His head circumference was normal. He doesn’t show broad thumbs and toes explain in RSTS (figure2b,2c); his stature was 162 CM so he did not show short stature as mention in RSTS. His affected brother has an intellectual disability and developmental delay and his healthy brother has a son with developmental delay but his family has no intention to apply genetic diagnosis for this child. His mother was healthy and his father passed away but according to his family he was normal.

Figure 1: Pedigree of the family with deletion in CREBBP gene.
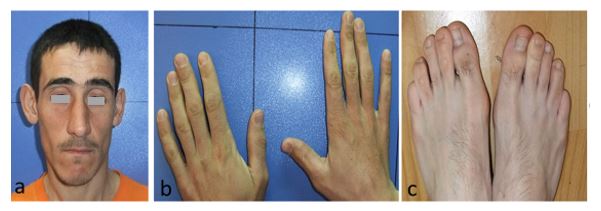
Figure 2: Picture of affected individual with deletion in CREBBP gene and SHANK3 gene. Ptosis and heavy eyebrow and highly set ears and normal thumbs and toes is recognizable.
Material and Methods
Cytogenetic analyses were performed by Gbanding techniques at 550 bands of resolution on metaphase chromosomes obtained by standard procedures from peripheral blood lymphocytes. Then whole blood DNA sample for MLPA testing were prepared using standard salting out method [12] and quantified by spectrophotometer (Nanodrop). Further investigation was performed using Multiplex Ligation Probe Amplification (MLPA) technique with P245 kit, P070 and P036 kits (SALSA, MLPA Kit, MRC, Holland) to screen micro-deletion syndromes and subtelomeric rearrangements. MLPA results were analyzed by Genemaker V1.95.
Results
Cytogenetic analysis of proband, his mother and affected brother were normal. Performing (MLPA) experiments shown micro-deletion in CREBBP gene (16p13.3) and micro-deletion in SHANK3 gene (22q13.33) in P245-kit but his affected brother has not shown any abnormality in these regions. Using another MLPA kit (p070) revealed a micro-deletion in DECR2 gene which located at chromosome 16p13.3 region in the proband, again his brother MLPA results in this kit was normal. MLPA results of proband as an electro-photogram has been shown in figure 3.
Discussion
The present study has been reported a family with affected child who has deletion in the CREBBP gene and miss some sign of classical RSTS especially broad thumb and toes, in the same time this patient has a deletion in the SHANK3 gene. The most patients with deletion in CREBBP or EP300 or both genes phenotypically show the RSTS signs especially broad thumb and toes but using next generation sequence in some patients with developmental delay or Intellectual disability represent individuals with deletion in these two genes and without these signs of RSTS [10].
Applying MLPA in our patient revealed deletion in CREBBP; the RSTS gene; and some signs of RSTS such as ptosis, speech difficulty, seizures and heavy eyebrows but other expecting symptom such as short stature and broad thumbs and toes has not detected. Menke in his report, after presenting 11 patients with CREBBP deletion and without RSTS sign, explained that phenotype variation has been expected because by using new techniques such as next generation sequencing the genetic cause of diseases has been revealed and genotypephenotype correlation could be explained according by these new information. Especially after the detection of exact genetic cause of syndromes, variability of symptoms or phenotypes could be expected [10]. In the other hand patients could have deletion in the special gene with or without some sign of diseases. In our case existence of some RSTS signs could be explain by deletion of CREBBP gene but there is some other deletion in our patient that might be the source of absence of some typical sign or presence of other sign which has not detected in the cases with RSTS before.

Figure 3: MLPA results using the MRC-Holland P245 kit. Deletion in CREBBP gene in position (16p13.3) and SHANK gene in position (22q13.33) are shown by arrow in GeneMarker electro-photogram.
Although deletion of EP300 gene has been detected in some patients with RSTS as well as CREBBP gene, none of them, in our knowledge, have been identified with deletion in SHANK3 gene. SHANK3 gene with 22 exons is located on chromosome 22 (22q13.3) and deletion of this gene has been known as Phelan-McDermid syndrome (PHMDS) (OMIM#606232) and mainly reported symptoms include dolichocephaly and macrocephaly, pointed chin, ptosis, delayed motor development, seizures, tall stature, delayed expressive speech Shank family are an important proteins at the postsynaptic density which involved in aspects of synaptic development and function [13], SHANK3 deletion has been detected in some other diseases such as bipolar disorder [14] and autism spectrum disorders [15].
Deletion in SHANK3 gene has been detected in our patient as well as deletion of CREBBP gene. We couldn’t find some sign of PHMDS such as dolichocephaly and macrocephaly, pointed chin and tall stature in our patient. Other sign such as ptosis, developmental delay, intellectual disability, seizures and delayed, expressive speech could be explained by both syndromes. Tall stature in PHMDS and short stature in RSTS has not detected may be the average stature of this patient is sum of these two syndrome.
MLPA technique has been revealed another deletion in our patient in chromosome at 16p13.3 region (GRCh38/hg38), representing DECR2 gene and in our knowledge has not been reported in the ID patients before. There is a large distance between these two deleted genes (3.3Mb) and although they located in the same band of chromosome 16, large deletion including both of them would be detected in conventional cytogenetic analysis and we could not detect such deletion in his karyotype. DECR2 deletion in the other hand could be a technical artifact and needs more investigation to detect effects of this gene in ID patients. SHANK3 and EP300 located in the same chromosome (22) as well but we could not recognize any kind of deletion in his conventional karyotype and need high resolution analysis or molecular cytogenetic to detect exact size of deletion.
We applied MLPA kits to his affected brother individual of this family but could not detect any deletion or addition. In his physical examination we just could detect intellectual disability, long face, beaked nose without broad thumb and toes. Genetic cause of intellectual disability in his brother would be different that we could not find in MLPA technique and needs to study more to detect the exact genetic cause of his intellectual disability but we could not apply any further technique because the family refused to cooperate. We believe with family cooperation, it is possible to reach new sight for this syndrome and as we know this is the first report of deletion in SHANK3 gene in patient with deletion of CREBBP gene without RSTS typical signs.
Acknowledgements
The authors extend their gratitude to the patients and their willingness to participate in this study.
Funding
This article is result of PhD thesis project that has been founded by Kawsar human genetic research center (KHGRC), Tehran, Iran.
Conflict of Interest
All authors hereby affirm that the manuscript is original, that all statements asserted as facts are based on authors careful investigation and accuracy, that the manuscript has not been published and has not been submitted or considered for publication elsewhere, and that authors have full power authority to enter into this copyright assignment and to make the grants herein content.
Ethical approval
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5).
Informed consent
Informed consent was obtained from the reporting patient guardian for being included in the study. Additional informed consent was obtained from all patients for whom identifying information is included in this article.
References
1. Park E, Kim Y, Ryu H, et al. 2014. Epigenetic mechanisms of Rubinstein-Taybi syndrome. Neuromolecular Med. 16: 16-24. Ref.: https://bit.ly/2zn1lsG
2. Nayak RB, Lakshmappa A, Patil NM, et al. 2012. Rubinstein-Taybi Syndrome with Psychosis. Indian J Psychol Med. 34: 184-186. Ref.: https://bit.ly/2zpV9jS
3. Munevveroglu AP, Akgol BB. 2012. Rubinstein-taybi syndrome: a case report. Case Rep Dent. 2012: 483867. Ref.: https://bit.ly/2zqbDIB
4. Kumar S, Suthar R, Panigrahi I, et al. 2012. Rubinstein-Taybi syndrome: Clinical profile of 11 patients and review of literature. Indian J Hum Genet. 18: 161-166. Ref.: https://bit.ly/324OFTJ
5. Stef M, Simon D, Mardirossian B, et al. 2007. Spectrum of CREBBP gene dosage anomalies in Rubinstein-Taybi syndrome patients. Eur J Hum Genet. 15: 843-847. Ref.: https://bit.ly/327yrtc
6. Roelfsema JH, White SJ, Ariyurek Y, et al. 2005. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 76: 572-580. Ref.: https://bit.ly/2zm7xkQ
7. Negri G, Magini P, Milani D, et al. 2016. From Whole Gene Deletion to Point Mutations of EP300-Positive Rubinstein-Taybi Patients: New Insights into the Mutational Spectrum and Peculiar Clinical Hallmarks. Hum Mutat. 37: 175-183. Ref.: https://bit.ly/2Nz0jlV
8. Milani D, Manzoni FM, Pezzani L, et al. 2015. Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Ital J Pediatr. 41: 4. Ref.: https://bit.ly/2HrG6uv
9. Van Belzen M, Bartsch O, Lacombe D, et al. 2011. Rubinstein-Taybi syndrome (CREBBP, EP300). Eur J Hum Genet. 19: 121. Ref.: https://bit.ly/2LeMWob
10. Menke LA, van Belzen MJ, Alders M, et al. 2016. CREBBP mutations in individuals without Rubinstein-Taybi syndrome phenotype. Am J Med Genet A. 170: 2681-2693. Ref.: https://bit.ly/2Zlw5Kf
11. Sellars EA, Sullivan BR, Schaefer GB. 2016. Whole exome sequencing reveals EP300 mutation in mildly affected female: expansion of the spectrum. Clin Case Rep. 4: 696-698. Ref.: https://bit.ly/2ZrUUn3
12. Jawdat N Gaaib. 2011. Simple salting – out method for genomic DNA extraction from whole blood. Tikrit Journal of Pure Science. 16: 9-11. Ref.: https://bit.ly/2KXhqfl
13. Halbedl S, Schoen M, Feiler MS, et al. 2016. Shank3 is localized in axons and presynaptic specializations of developing hippocampal neurons and involved in the modulation of NMDA receptor levels at axon terminals. J Neurochem. 137: 26-32. Ref.: https://bit.ly/2PfHHtG
14. Zhang C, Wu Z, Hong W, et al. 2016. Evaluating the association between the SHANK3 gene and bipolar disorder. Psychiatry Res. 244: 284-288. Ref.: https://bit.ly/30AWnVv
15. Tao-Cheng JH, Toy D, Winters CA, et al. 2016. Zinc Stabilizes Shank3 at the Postsynaptic Density of Hippocampal Synapses. PLoS One. 11: Ref.: https://bit.ly/2NADiiy


















