
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Short CommunicationDOI Number : 10.36811/ijgmgt.2020.110005Article Views : 11Article Downloads : 9
The p53 Slavic Gene Mutation Nijmegen Breakage Syndrome
Saptarshi Pal1* and Elena Ponomarenko2
1Scientific advisor: PhD, Department of Pathophysiology, Perm State Medical University named after academician E.A. Wagner, Perm, Russia
2Associate Professor, Department of Pathophysiology, Perm State Medical University named after academician E.A. Wagner, Perm, Russia
*Corresponding Author: Saptarshi Pal, Scientific advisor: PhD, Department of Pathophysiology, Perm State Medical University named after academician E.A. Wagner, Perm, Russia, Email: bpalsas@gmail.com
Article Information
Aritcle Type: Short Communication
Citation: Saptarshi Pal, Elena Ponomarenko. 2020. The p53 Slavic Gene Mutation Nijmegen Breakage Syndrome. Int J Genet Med Gene Ther. 2: 01-05.
Copyright: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2020; Saptarshi Pal
Publication history:
Received date: 20 January, 2020Accepted date: 18 February, 2020
Published date: 20 February, 2020
Abstract
Nijmegen breakage syndrome is a rare autosomal congenital disorder. It originates from mutations in the NBS1 gene located in band 8q21, which encodes for the DNA double strand repair protein Nibrin. Immunodeficiency with increased susceptibility to infection. It is a condition characterized by short stature, an unusually small head size (microcephaly), distinctive facial features, recurrent respiratory tract infections, an increased risk of cancer, intellectual disability, and other health problems. People with this condition typically grow slowly during infancy and early childhood. After this period of slow growth, affected individuals grow at a normal rate but remain shorter than their peers. Microcephaly is apparent from birth in the majority of affected individuals. The head does not grow at the same rate as the rest of the body, so it appears that the head is getting smaller as the body grows (progressive microcephaly).Individuals with Nijmegen breakage syndrome have distinctive facial features that include a sloping forehead, a prominent nose, large ears, a small jaw, and outside corners of the eyes that point upward (up slanting palpebral fissures). These facial features typically become apparent by age 3.
Keywords: Nijmegen breakage syndrome; Chromosomal instability; Immunodeficiency; Microcephaly
Epidemiology: Nijmegen breakage syndrome is a rare disease and there are no reliable estimates of its prevalence. The largest groups of patients were West Slavic origins diagnosed in Poland, Czech Republic, Ukraine and some parts of Russia. The number of known patients identified worldwide increased significantly when the disease-causing gene, NBN, was identified. Apart from over 150 subjects reported in the medical literature [12-36], there are many more patients recorded in national registries (e.g. Czech and Polish- CKH p.c.). Currently, the largest European registry is managed by the European Society for Immunodeficiencies (ESID). The disease seems to occur worldwide, but with a distinctly higher prevalence among Central European and Eastern European populations, i.e. in the Czech Republic, Poland, Russia and Ukraine. The proportion of patients identified in these populations correlates with a high carrier frequency of the major NBN mutation, c.657_661del5, estimated to be 1 case per 177 newborns, clearly the result of a founder effect. Nijmegen breakage syndrome has also been reported in many other European countries as well as in North and South America, Morocco and New Zealand.
Diagnosis: Nijmegen breakage syndrome (NBS) should be suspected in individuals with the following clinical and supportive laboratory findings.
Clinical features
• Disproportionate microcephaly that is progressive
• Craniofacial features that include a sloping forehead, upward slanted palpebral fissures, prominent nose, relatively large ears, and retrognathia
• Growth retardation that is more pronounced from birth until the age of three years, with mild improvement thereafter
• Recurrent infections including pneumonia, bronchitis, sinusitis, otitis media, and mastoiditis
• Malignancies, predominantly of lymphoid origin
• Decline in intellectual ability, from normal or borderline-normal during early childhood to moderate intellectual disability in older individuals
Supportive laboratory findings
• Immunodeficiency o Severe hypogammaglobulinemia
o Deficiencies of IgG2 and IgG4 are frequent even when the IgG serum concentration is normal.
o The most commonly reported defects in cellular immunity include reduced absolute numbers of total B cells, CD3+ T cells, and CD4+ cells
o An increased frequency of T cells with a memory phenotype (CD45RO+) and a concomitant decrease in naïve T cells (CD45RA+).
o The in vitro proliferation of T and B lymphocytes to antigen and/or mitogenic stimuli is greatly reduced in most affected individuals.
• Chromosome instability
o Inversions and translocations involving chromosomes 7 and 14.
o The breakpoints most commonly involved are 7p13, 7q35, 14q11, and 14q32, which are the loci for immunoglobulin and T cell-receptor genes.
• Radiation- Cells from individuals with NBS have a decrease in colony-forming ability following exposure to ionizing radiation and radio mimetics in vitro.
Note: This test requires that a lymphoblastoid cell line be established. Because this process is more commonly performed in a research lab than in a clinical lab, the test may not be widely available clinically.
Single gene testing
• Targeted analysis for the pathogenic variant c.657_661del5 can be performed first. The c.657_661del5 pathogenic variant is detected in:
o ~100% of alleles in individuals of Slavic (Poland, Czech Republic, Ukraine) ancestry;
o ~70% of alleles in individuals of North American ancestry.
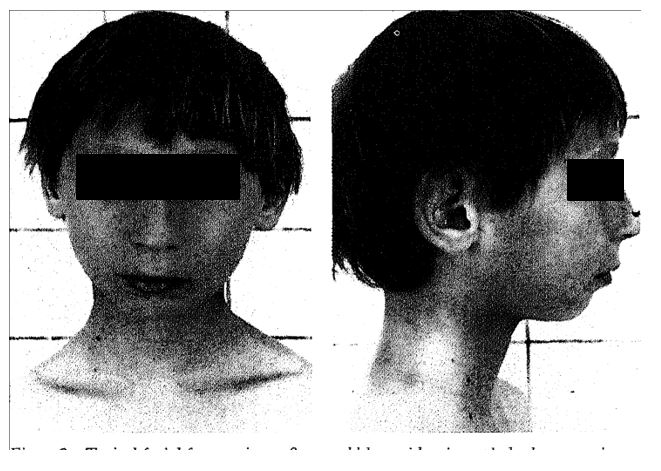
Typical facial features in an 8-year-old boy with microcephaly, long prominent nose, and small chin.
Treatment: No specific therapy is available for NBS. Due to the specific basic defect underlying immunodeficiency and sensitivity to IR, patients with NBS require multidisciplinary medical management and long-term follow-up. Specialized care provided by clinicians aware of these problems and of the natural history of the disease can prevent some complications, avoid unnecessary and excessive exposition to IR and take into consideration the high risk for malignancy. MRI and ultrasound examination are recommended as imaging techniques rather than CT scan or X-ray. It is important that patients with NBS are under the care of one primary physician, preferably a pediatrician or a general practitioner, who is acquainted with the condition. Systematic (prophylactic) supervision by an immunologist and oncologist is recommended. Female patients should be under the care of an endocrinologist and a gynecologist when they reach pubertal age. Psychological, social and educational support is essential for improvement of the quality of patients’ life.
Substitution with immunoglobulins (i.e., [IVIG] therapy) is indicated in patients with agammaglobulinemia and in children with IgG2 deficiency. Before IVIG is started, the presence of anti-IgA antibodies must be determined in patients with IgA deficiency. Consider antibiotic prophylaxis in patients with recurrent respiratory tract infections. Urinary tract infections due to congenital malformations of the kidneys occur in some children; antibiotic prophylaxis is indicated in these patients. The intensity of therapy must be adapted to individual risk factors and tolerance. Anthracycline-induced cardiomyopathy was reported in one patient, and, therefore, echocardiographic monitoring is strongly recommended. Bone marrow transplantation (BMT) or hematopoietic stem cells transplantation (HSCT) is an option that can be considered in some patients with Nijmegen breakage syndrome. When hypergonadotropic hypogonadism is confirmed, substitution hormone therapy to support the development of secondary sex characteristics and to prevent osteoporosis must be considered when the patient reaches the appropriate age.
Vitamin E supplementation in doses appropriate for age and body weight is recommended, as in individuals with other chromosome instability disorders.
Conclusion
Because patients with NBS have an increased risk of developing malignancies, it is important to diagnose this disease at an early age. In NBS patients x irradiation should be avoided as much as possible, for therapeutic but also for diagnostic reasons. In all patients with unexplained early progressive microcephaly and growth retardation, chromosome instability analysis should be performed, in order to establish or exclude a chromosome instability disorder.
References
1. Weemaes CMR, Hustinx TWJ, Scheres JMJC, et al. 1981. A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr Scand. 70: 557- 564. Ref.: https://bit.ly/39CnJyq
2. Seemanova E, Passarge E, Beneskova D, et al. 1985. Familial microcephaly with normal intelligence, immunodeficiency and risk for lymphoreticular malignancies: a new autosomal recessive disorder. Am J7 Med Genet. 20: 639-648. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/3857858
3. Chrzanowska KH, Kleijer WJ, Krajewska- Walasek M, et al. 1995. Eleven Polish patients with microcephaly, immunodeficiency and chromosomal instability; the Nijmegen breakage syndrome. Am J Med Genet. 57: 462-471. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/7545870
4. Demuth I, Digweed M. 2007. The clinical manifestation of a defective response to DNA double-strand breaks as exemplified by Nijmegen breakage syndrome. Oncogene. 26: 7792-7798. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/18066092
5. Pluth JM, Yamazaki V, Cooper BA, et al. 2008. DNA double-strand break and chromosomal rejoining defects with misrejoining in Nijmegen breakage syndrome cells. DNA Repair (Amst). 7: 108-118. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/17919995
6. Maraschio P, Peretti D, Lambiase S, et al. 1986. A new chromosome instability disorder. Clin Genet. 30: 353-365. Ref.: https://bit.ly/2uTZ1uO
7. Curry CJR, O'Lague P, Tsai J, et al. 1989. AT(Fresno): a phenotype linking ataxia-telangiectasia with the Nijmegen breakage syndrome. Am J Hum Genet. 45: 270-275. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/2491181
8. Jaspers NGJ, Gatti RA, Baan C, et al. 1988. Genetic complementation analysis of ataxia telangiectasia and Nijmegen breakage syndrome: a survey of 50 patients. Cytogenet. Cell Genet. 49: 259- 263. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/3248383
9. Cerosaletti KM, Lange E, Stringham HM, et al. 1998. Fine localization of the Nijmegen breakage syndrome gene to 8q21: evidence for a common founder haplotype. Am J Hum Genet. 63: 125-134. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/9634525
10. Cheung VG, Ewens WJ. 2006. Heterozygous carriers of Nijmegen breakage syndrome have a distinct gene expression phenotype. Genome Res. 16: 973-979. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/16809669
11. Conley ME, Spinner MB, Emanuel BS, et al. 1986. A chromosome breakage syndrome with profound immunodeficiency. Blood. 67: 1251-1256. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/2421804
12. Curry CJR, O'Lague P, Tsai J, et al. 1989. AT(Fresno): a phenotype linking ataxia-telangiectasia with the Nijmegen breakage syndrome. Am J Hum Genet. 45: 270-275. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/2491181
13. Der Kaloustian VM, Kleijer W, Booth A, et al. 1996. Possible new variant of Nijmegen breakage syndrome. Am J Med Genet. 65: 21-26. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/8914736
14. Komatsu K, Matsuura S, Tauchi H, et al. 1996. The gene for Nijmegen breakage syndrome (V2) is not located on chromosome 11. (Letter) Am J Hum Genet. 58: 885-888. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/8644753
15. Meyer S, Kingston H, Taylor AMR, et al. 2004. Rhabdomyosarcoma in Nijmegen breakage syndrome: strong association with perianal primary site. Cancer Genet Cytogenet. 154: 169-174. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/15474156
16. Saar K, Chrzanowska KH, Stumm M, et al. The gene for the ataxia-telangiectasia variant, Nijmegen breakage syndrome, maps to a 1-cM interval on chromosome 8q21. Am J Hum Genet. 60: 605-610. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/9042920
17. Taalman RDFM, Hustinx TWJ, Weemaes CMR, et al. 1989. Further delineation of the Nijmegen breakage syndrome. Am J Med Genet. 32: 425-431. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/2786340
18. Teebi AS, Al-Awadi SA, White AG. 1987. Autosomal recessive nonsyndromal microcephaly with normal intelligence. Am J Med Genet. 26: 355- 359. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/3812587
19. Tekin M, Dogu F, Tacyildiz N, et al. 657del5 mutation in the NBS1 gene is associated with Nijmegen breakage syndrome in a Turkish family. Clin Genet. 62: 84-88. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/12123493
20. van der Burgt I, Chrzanowska KH, Smeets D, et al. 1996. Nijmegen breakage syndrome. J Med Genet. 33: 153-156. Ref.: https://adc.bmj.com/content/82/5/400.short


















