
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Case ReportDOI Number : 10.36811/ijho.2020.110007Article Views : 7Article Downloads : 7
A Rare Case of Portal Vein Thrombosis due to Protein S deficiency in a patient with Decompensated Cryptogenic CLD with Portal Hypertension, Completely Recanalized by Single Oral Anticoagulant-Rivaroxaban
Richmond Ronald Gomes
Associate Professor, Medicine, Ad-din Women’s Medical College Hospital, Dhaka, Bangladesh
*Corresponding Author: Dr. Richmond Ronald Gomes, Associate Professor, Medicine, Ad-din Women’s Medical College Hospital, Dhaka, Bangladesh, Tel: +8801819289499; Email: rrichi.dmc.k56@gmail.com
Article Information
Aritcle Type: Case Report
Citation: Richmond Ronald Gomes. 2020. A Rare Case of Portal Vein Thrombosis due to Protein S deficiency in a patient with Decompensated Cryptogenic CLD with Portal Hypertension, Completely Recanalized by Single Oral Anticoagulant-Rivaroxaban. Int J Hematol Oncol. 3: 29-37.
Copyright: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2020; Richmond Ronald Gomes
Publication history:
Received date: 10 August, 2020Accepted date: 10 September, 2020
Published date: 12 September, 2020
Abstract
Venous thromboembolic diseases are a group of heterogeneous diseases with different clinical forms and prognosis. Abdominal venous thrombosis may present either as Budd-Chiari syndrome (BCS) caused by hepatic vein or proximal inferior vena cava (IVC) obstruction or as an extra hepatic portal obstruction (EHPVO) caused by Portal vein thrombosis or mesenteric vein thrombosis. Portal vein thrombosis (PVT) is a rare form of venous thrombosis that affects the hepatic portal vein flow, which can lead to portal hypertension. Treatment of PVT includes anticoagulants, thrombolysis, and insertion of shunts, bypass surgery, and liver transplantation. Single anticoagulation therapy can be associated with a reduction in new thrombotic episodes. Here we experienced a 23 year old young lady with history of recent intrauterine death (IUD) diagnosed as PVT provoked by protein S deficiency with newly diagnosed decompensated cryptogenic chronic liver disease with portal hypertension. PVT was completely recanalized with single oral anticoagulant therapy rivaroxaban as initial low molecular weight heparin, enoxaparin administration caused reversible pancytopenia and there is a concern for bleeding and regular monitoring of INR with warfarin in this patient.
Keywords: Portal vein thrombosis; Chronic liver disease; Protein S deficiency; Oral anticoagulant; Portal hypertension; Thrombolysis
Introduction
Portal vein thrombosis (PVT) is defined as complete or partial obstruction of blood flow in the portal vein, associated with a thrombus in the vessell lumen [1]. The first case of PVT was reported in 1868 by Balfour and Stewart, in a patient showing splenomegaly, ascites, and variceal dilatation [2]. PVT is rare in the general population having been reported with mean age-standardized incidence and prevalence rates of 0.7 and 3.7 per 100000 inhabitants, respectively [3]. However among patients with cirrhosis, these rates jump to between 4.4%-15%, and cause about 5%-10% of overall cases of portal hypertension [4]. Some 22%-70% of patients without cirrhosis demonstrate prothrombotic states and local factors are present in 10%-50% [3-5], although more than one factor is often identified [6]. PVT also shows different clinical presentations in acute vs chronic onset patients. It causes portal hypertension and varix, and if severe, can cause variceal hemorrhage and ischemic bowel disease [7] PVT is classified into four categories: (1) thrombosis confined to the portal vein beyond the confluence of the splenic and superior mesenteric vein (SMV); (2) extension of thrombus into the SMV, but with patent mesenteric vessels; (3) diffuse thrombosis of splanchnic venous system, but with large collaterals; and (4) extensive splanchnic venous thrombosis, but with only fine collaterals. Currently this anatomical classification is mainly used to determine operability, In all cases, patients with PVT should be tested for an underlying thrombophilic condition [6]. Hereditary thrombophilia’s known to predispose for PVT include mutations of the prothrombin, or factor V, genes, and deficiency of one of the natural anticoagulant proteins C, S, or antithrombin III. Fisher et al [8], in a study with twenty-nine adult patients with portal hypertension caused by PVT, found that 18 patients (62%) had deficiencies in one or more of the natural anticoagulant proteins, and six had combined deficiency of all three proteins. Of these, eight cases (28%) had combined C and S protein deficiency, nine (31%) had C protein and antithrombin deficiency, seven (24%) showed protein S and antithrombin deficiency, and six cases (21%), as mentioned, had combined deficiency of all three proteins. We report a case of PVT caused by protein S deficiency that ended in successful reperfusion with single oral anticoagulation therapy by rivaroxaban.
Case presentation
A 23 year old nonalcoholic, non-smoker lady (gravida 2), not known to have diabetes and hypertension with the history of recent IUD due to PIH(pregnancy induced hypertension) at 35+ weeks of gestation referred from department of gynecology and obstetrics presented to us with the complaints of progressive abdominal distention for 15 days and upper abdominal pain for 3 days. But she denied any history of yellow coloration of urine and sclera, shortness of breath, decrease in urinary output, altered sensorium involuntary movement, fever, joint pain, any pigmentation hematemesis or melena. Her bowel habit was normal. Apart from calcium and hematinic, she was free of any medications. There was neither personal nor family history of liver disease nor any venous thrombosis. She had no past history of jaundice or previous fetal loss or taking any oral contraceptives prior pregnancy. On examination, she was conscious, oriented. Mild pallor was present but there was no jaundice or cyanosis. There were no skin or nail changes including no spider angioma. Axillary hair was sparse. Mild pedal edema was present. Vitals were stable. Flapping tremor was absent. Abdominal examination revealed hugely distended abdomen with presence of abdominal striae and distended vein the flow of which suggests portal venous obstruction. Ascites was present as suggested by presence of fluid thrill. Organomegaly could not ascertained due to tense ascites. But there was tenderness over right hypochondriac region without any rebound tenderness. Other systemic examination revealed no abnormalities. Complete blood count showed mild normocytic normochromic anemia with hemoglobin 10.3 gm/dl(normal 12-16 gm/dl), MCV 81.4 fL(normal 78-98 fL), MCH 28.5 pg(normal 26-32 pg), total white cell count 7.61×103/uL (N-67.3%, L-18.9%), total platelet count 152×103/uL (150-450×103/uL). Serum bilirubin 1.1 mg/dl (normal 0.5-1.8 mg/dl) SGPT- 24 U/L (normal up to 31U/L), serum total protein 44.89g/L (normal 66-83 g/L), serum albumin 14.15 g/L (normal 35-52 g/L), serum globulin 30.74 g/L (normal 23-35 g/L), A: G 0.46:1 (normal 1.1:1), serum creatinine 0.68 mg/dl (normal 0.5-1.3 mg/dl). Serum electrolyte showed sodium 139 mmol/L (normal 135-145 mmol/L), potassium 3.8 mmol/L (normal 3.5-5.5 mmol/L). ANA was negative (17.51 U/ml, normal < 20 U/ml). TSH was mildly raised 8.42 mIU/mL (normal 0.35-5.5 mIU/mL).Prothrombin time was normal with 1 second difference between patient and control with INR 0.96.D-dimer values were more than 200 micro gm/ml.Urine routine examination showed ++proteinuria with presence of 8% (non-significant) dysmorphic RBC with urinary total protein 1.96g/24 hours. Ascitic fluid study showed lymphocyte rich (total cell 350/cmm, L-99%) transudative ascites (protein 0.29 g/dl). Ascitic fluid ADA was 2.55 U/L (normal <25 U/L). Doppler ultrasonography of whole abdomen revealed early chronic parenchymal change of liver with widened portal vein (15.4 mm) suggestive of portal vein thrombosis, splenomegaly with mildly dilated portal vein, postpartum grossly bulky uterus, huge ascites (Figure 1). Echocardiography failed to reveal any cardiac disease. HBsAg, Anti HBc (total), Anti HCV were negative. Urinary copper excretion was insignificant (12.30 mg/dl, normal 0-105 mg/dl). Thrombophillia screen was performed,not including screening for JAK2V617F mutation, homocysteine level, prothrombin mutation, factor V Leiden mutation revealed negative lupus anticoagulant, anti cardiolipin antibody. Anti thrombin III (106%, normal 75-125%) and protein C (54%, normal 50-120%) were normal but protein S level was significantly reduced (34%, normal 50-120%). Endoscopy of upper GIT, liver biopsyand fibroscan of liver could not be done due to COVID situation. On the basis of clinical features and lab parameters she as diagnosed as a case of decompensated cryptogenic chronic liver disease with portal hypertension with portal vein thrombosis secondary to protein S deficiency. So she was started treatment with large volume paracentesis with intravenous albumin support, diuretics, low dose thyroxin, nonselective beta blocker, and subcutaneous therapeutic doses of enoxaparin along with fluid and salt restriction. With treatment she started responding both clinically and biochemically. But on 3rd day of anticoagulation, repeat complete blood count revealed pancytopenia (anemia with hemoglobin 8.1 gm/dl(normal 12-16 gm/dl), MCV 86.7 fL (normal 78-98 fL), MCH 27.3 pg(normal 26-32 pg), total white cell count 2.25×103/uL (N-71.6%, L-14.8%), total platelet count 70×103/uL (150-450×103/uL). Peripheral blood film showed pancytopenia with negative direct coomb’s test, Serum reticulocyte count was reduced 0.50% (normal 1.5%-2.5%). As there was a suspicion of enoxaparin induced pancytopenia, enoxaparin was substituted with tablet rivaroxaban. Daily monitoring of complete blood count was done which revealed progressive improvement of count in all cell lines. During discharge, 7th day after stopping enoxaparin, her complete blood count showed hemoglobin 8.4 gm/dl(normal 12-16 gm/dl), MCV 84.5 fL (normal 78-98 fL), MCH 26.5 pg (normal 26-32 pg), total white cell count 3.27×103/uL(N-54.8%, L-36.7%), total platelet count 145×103/uL(150-450×103/uL). After 2 weeks during outpatient door follow up her complete blood count revealed almost normal count of all cell lines with hemoglobin 11.10 gm/dl(normal 12-16 gm/dl), MCV 85.1 fL(normal 78-98 fL), MCH 28.1 pg(normal 26-32 pg), total white cell count 6.10×103/uL(N-67%, L-26%), total platelet count 152×103/uL(150-450×103/uL). Rivaroxaban was continued for six months without any complications. After 6 months repeat Doppler ultrasonography of abdomen showed complete recanalization of portal vein with improved flow with normal portal venous diameter (Figure-2). There was no ascites.
Figure 1: Doppler ultrasound showed the thrombus in the intrahepatic portal vein and the widening of the portal vein diameter. (B) There is echogenic material and flow in the right portal vein lumen and multiple collateral vessels are not shown on the porta hepatis.
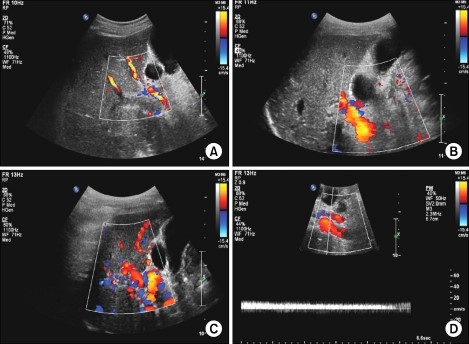
Figure 2: Doppler ultrasound showed (C) There is no echogenic material in the right portal vein lumen and the flow can be observed. (D) The flow and Doppler wave are noted in the umbilical portion of the left portal vein.
Discussion
Abdominal venous thrombosis is a rare disorder. It presents either as Budd-Chiari syndrome (BCS) or splanchnic vein thrombosis (SVT), but a mixed involvement is uncommon. PVT interrupts blood flow to the liver and causes portal hypertension [6]. Phlebothrombosis is caused by local, genetic, or acquired thrombophilic factors [9]. Local factors include local inflammatory lesions such as neonatal funiculitis, diverticulitis, appendicitis, pancreatitis, duodenal ulceration, tuberculous lymphadenitis, portal vein injury by surgical operations, thrombokinesis, pressure mass effect by an abdominal neoplasm, and portal hypertension by liver cirrhosis [9]. Denninger et al. reported that 6% to 11% of PVT is attributed to liver cirrhosis, and 35% is related to hepatocellular carcinoma [6]. Webb et al. reported that 40% of PVT is caused by intraperitoneal or systemic septicemia. In other reports, 7% of PVT was reported to be caused by chronic pancreatitis [10]. Primary myeloproliferative disorder, antiphospholipid antibody syndrome, paroxysmal nocturnal hemoglobinuria, oral contraceptives, pregnancy, childbirth, malignant tumor, and hypercysteinemia are reported as acquired thrombophilic factors [9]. Genetic thrombophilic factors can be divided into the frequent type, such as factor V Leiden mutation and mutation IIG20210, in which PVT is rarely associated, and the rare type, such as protein C, S, and antithrombin III, in which PVT is more likely to occur [9].Among hereditary risk factors, Factor V Leiden mutation is common in BCS and protein C deficiency is common in portal vein thrombosis (PVT) [11]. Deficiency of protein C is more common than protein S deficiency in both BCS and PVT [12]. However, protein C deficiency is present in approximately 2%-5% patients presenting VTE. Severe homozygous or compound heterozygous protein C deficiency is found in 1 in 500000-750000 live births. Protein S deficiency occurs in 1.35% of the patients with venous thrombosis.The level of protein S was low and that of protein C was just below the normal level in the present case.There is evidence to suggest that thrombosis in unusual sites, such as cerebral sinus venous thrombosis, mesenteric vein thrombosis, PVT, and suprahepatic vein thrombosis (Budd-Chiari syndrome), in young individuals is associated with inherited thrombophilia. Inherited thrombophilia is found in patients with SVT, although making diagnoses of inherited deficiencies of antithrombin, protein C, and protein S is difficult in the presence of liver impairment, which causes a reduced synthesis of the natural anticoagulant proteins [3,13-15]. This difficulty is not seen with the Factor V Leiden and prothrombin G20210A mutations. D-dimer is considered as a marker of hypercoagulable state, besides endogenous fibrinolysis and so, increased D-dimer levels are detectable in patients who are affected by arterial and/or venous thrombosis, as it was seen in the present case [16]. In Mexico, Majluf-Cruz et al [17], studied 36 patients who had thrombosis-related portal hypertension and found an incidence of 30% of protein C deficiency, whereas 9% had protein S deficiency in patients with primary thrombophilia [18]. Similarly in Mexican patients with non-cirrhotic PVT, 31% had protein C deficiency [19]. However, a French study has found a high number of patients with non-cirrhotic PVT showed Protein S deficiency [20] and in a study from United Kingdom, protein S deficiency was found in 38% of patients with PVT [21]. Other cases have also reported C and S protein deficiencies in patients with idiopathic portal hypertension accompanied by PVT [22,23]. Valla et al [20], argue that C and S protein deficiencies do not explain the majority of idiopathic portal thrombosis. Nevertheless, we agree with others that measurements of C and S proteins should be performed in patients with portal thrombosis when no overt cause is located. However, since a low number of cases of PVT may be due to underlying hereditary anticoagulant protein deficiency, this can only be confirmed by careful investigation of background of family members, preferably including both parents. When studies of the parents is not feasible, another possibility might be screening siblings, which could be used for both diagnostic and counseling purposes. Lastly, the recent use of gene sequencing in the elucidation of anticoagulant protein gene mutations may now allow determination of whether such anticoagulant deficiencies in PVT are truly primary or not [24]. Some possible mechanisms for reduction in concentrations of procoagulant and anticoagulant proteins in patients with PVT are
? Hereditary or acquired thrombophilia
? Reduced hepatic blood flow
? Reduced synthesis
? Portal hypertension
? Portosystemic shunting
? Clearance or consumption
? Portal pyaemia or other local inflammatory disease
? Portal vein thrombosis
? Reduced levels of procoagulant and anticoagulant proteins
Clinically, PVT is classified as acute or chronic. However, it is difficult to identify when the symptoms begin and even the temporal criteria about chronicity have not yet been established [25]. Malkowski et al. classified PVT as acute or chronic according to symptom onset time before admission (60 days) [25]. Acute PVT typically provokes stomachache, nausea, emesis, and fever. The symptoms can be more severe if mesenteric infarction provokes infarction of the bowel or liver. Nonspecific symptoms such as diarrhea, anorexia, weight loss, and abdominal distention may develop [26]. The symptoms of chronic PVT are similar to the acute ones, but such symptoms may be related to portal hypertension and splenomegaly [27]. Splenomegaly, anemia, leukopenia, thrombocytopenia, and variceal bleeding can be provoked [27]. Our case developed signs of portal hypertension both clinically and on ultrasound imaging. Duplex, Doppler ultrasound, and abdominal CT scan can be used to make a accurate diagnosis of PVT in the early stage, and abdominal MRI can be another diagnostic tool [28]. In particular, abdominal MRI is known to be more useful than Doppler ultrasound in identifying venous collateral development and cavernoma [28]. In patients with PVT, local factors such as liver cirrhosis and pancreatitis should be suspected. If no local factor can be identified, it is necessary to investigate genetic or acquired thrombophilic factors [26]. TE is a non-invasive technique to assess liver fibrosis, which assesses liver fibrosis by calculating the velocity of a low-frequency transient shear wave produced by a mechanical probe that is placed directly on the skin of the patient. Liver stiffness is expressed in kPa. The method is easy to learn (the procedure can be performed by a technical assistant), and results are immediately available. One meta-analysis evaluating the predictive performance of TE in patients with chronic liver disease suggests the optimal cut-off value for the diagnosis of significant fibrosis is 7.65 kPa and for cirrhosis 13.01 kPa [29]. There is no data on the use of TE in PVT, but this method may be useful to determine liver fibrosis in these patients. Complications during follow-up frequently include: esophageal and gastric varices, portal hypertensive gastropathy and bleeding. Portal hypertensive gastropathy is reported to be 44% in patients without cancer and cirrhosis, as was the case with our patient [30]. Therefore, it would be wise to screen all PVT patients endoscopically. Although spontaneous resolution of PVT has been reported in the literature, a specific therapeutic management strategy is necessary.In our case, it was possible to infer that PVT was due to protein S deficiency either secondary to liver cirrhosis or due to inherited thrombophilia.The goal of treatment is similar in acute and chronic PVT, and includes correction of causal factors, prevention of thrombosis extension and achievement of portal vein patency. Currently, anticoagulant therapy is the best way to obtain portal vein recanalization; however, its application is not universally accepted. No controlled trial has been performed on the use of anticoagulants in acute PVT [31]. After 6 months of therapy, complete recanalization has been reported in about 50% of patients, with good outcomes in mesenteric vein involvement, and very few complications. What is certain is that, in acute PVT onset, the sooner the treatment is given, the better the prognosis; the rate of recanalization is about 69% if anticoagulation is begun within the first week after diagnosis, while it falls to 25% when begun in the second week [32]. Thrombolytic therapy may also be effective, but efficacy is significantly lower and mortality increases compared to conservative treatment [25]. Surgical thrombectomy is usually not recommended. Other approaches, such as transyugular intrahepatic portosystemic shunt, should be reserved for liver transplant patients developing acute PVT or as an alternative when anticoagulation fails [33]. In non-cirrhotic and non-neoplastic patients, PVT has shown promising results with overall survival at 1 year and 5 years of 92% and 76% respectively [3,30,34,35]. In many cases, chronic PVT is accompanied by gastroesophageal varix, which can result in variceal bleeding during anticoagulation therapy [26] In the case of acute PVT, however, early anticoagulant therapy can prevent the spread of thrombi and promote blood reperfusion [27]. However, there are no clear guidelines on the choice of anticoagulant drugs, the period of administration, and even the need for anticoagulant therapy [25]. In our case, we successfully treated our acute PVT patient with 6 months single oral anticoagulant therapy with rivaroxaban as initial administration of LMWH enoxaparin caused reversible pancytopenia and there is a concern for bleeding and regular monitoring of INR with warfarin in this patient.
Conclusion
In conclusion, our case shows that PVT can be provoked by protein S deficiency and that the PVT can be recanalized by short-term oral rivaroxaban therapy. Although the evidence is not definitive, existing literature supports the idea that the risk-benefit ratio favors anticoagulation in cirrhotic PVT. Further study is needed to investigate the period of administration, and the dosage and duration for relapse prevention in treatment with oral anticoagulants.
References
1. Kocher G, Himmelmann A. 2005. Portal vein thrombosis (PVT): a study of 20 non-cirrhotic cases. Swiss Med Wkly. 135: 372-376. Ref.: shorturl.at/lst15
2. Wang JT, Zhao HY, Liu YL. 2005. Portal vein thrombosis. Hepatobiliary Pancreat Dis Int. 4: 515-518.
3. Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al. 2010. Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology. 51: 210-218. Ref.: shorturl.at/gpJQ6
4. Ponziani FR, Zocco MA, Campanale C, et al. 2010. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol. 16: 143-155. Ref.:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2806552/
5. Janssen HL, Wijnhoud A, Haagsma EB, et al. 2001. Extrahepatic portal vein thrombosis: aetiology and determinants of survival. Gut. 49: 720-724. Ref.: shorturl.at/npLUZ
6. Denninger MH, Chaït Y, Casadevall N, et al. 2000. Cause of portal or hepatic venous thrombosis in adults: the role of multiple concurrent factors. Hepatology. 31: 587-591. Ref.: shorturl.at/qAPRU
7. Sheen CL, Lamparelli H, Milne A, et al. 2000. Clinical features, diagnosis and outcome of acute portal vein thrombosis. QJM. 93: 531-534. Ref.: shorturl.at/tzBG9
8. Fisher NC, Wilde JT, Roper J, et al. 2000. Deficiency of natural anticoagulant proteins C, S, and antithrombin in portal vein thrombosis: a secondary phenomenon? Gut. 46: 534-539. Ref.: shorturl.at/fsvHQ
9. Cheun JY, Lee TH, Kim YS, et al. 2008. Portal and splenic vein thrombosis successfully treated with low molecular weight heparin in acute pancreatitis. Korean J Med. 74: 37-41.
10. Sakorafas GH, Sarr MG, Farley DR, et al. 2000. The significance of sinistral portal hypertension complicating chronic pancreatitis. Am J Surg. 179: 129-133. Ref.: shorturl.at/giwQ1
11. Mohanty D, Shetty S, Ghosh K, et al. 2001. Hereditary thrombosis as a cause of Budd- Chiari syndrome: a study from western India . Hepatology. 34: 666-670. Ref.: shorturl.at/qKV58
12. Bhattacharyya M, Makharia G, Kannan M, et al. 2004. Inherited prothrombotic defects in Budd-Chiari syndrome and portal vein thrombosis: a study from North India. Am J Clin Pathol. 121: 844-847. Ref.: shorturl.at/cnKL9
13. Darvish Murad S, Plessier A, Hernandez-Guerra M, et al. 2009. EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd- Chiari syndrome. Ann Intern Med. 151: 167-175. Ref.: shorturl.at/gmBR0
14. Martinelli I, Primignani M, Aghemo A, et al. 2009. High levels of factor VIII and risk of extra-hepatic portal vein obstruction. J Hepatol. 50: 916-922. Ref.: shorturl.at/jBOP0
15. Dentali F, Galli M, Gianni M, et al. 2008. Inherited thrombophilic abnormalities and risk of portal vein thrombosis. A metaanalysis. Thromb Haemost. 99: 675-680. Ref.: shorturl.at/sAXY9
16. Perrier A, Bounameaux H. 2001. Cost-effective diagnosis of depp vein thrombosis and pulmonary embolism. Thromb Haemost. 86: 475-487. Ref.: shorturl.at/fpOY3
17. Majluf-Cruz A, Hurtado Monroy R, Sansores García L, et al. 1996. The incidence of protein C deficiency in
thrombosis-related portal hypertension. Am J Gastroenterol. 91: 976-980. Ref.: shorturl.at/flvU1
18. Ruiz-Argüelles GJ, López-Martínez B, Valdés-Tapia P, Gómez. et al. 2005. A comprehensive prospective study indicates that most cases are multifactorial. Am J Hematol. 78: 21-26. Ref.: shorturl.at/tvQW5
19. Orozco H, Guraieb E, Takahashi T, Garcia, et al. 1988.Deficiency of protein C in patients with portal vein thrombosis. Hepatology. 8: 1110-1111. Ref.: shorturl.at/ewxzQ
20. Valla D, Casadevall N, Huisse MG, et al. 1988. Etiology of portal vein thrombosis in adults. A prospective evaluation of primary myeloproliferative disorders. Gastroenterology. 94: 1063-1069.
21. Aiach M, Borgel D, Gaussem P, et al. 1997. Protein C and protein S deficiencies. Semin Hematol. 34 : 205-216. Ref.: https://pubmed.ncbi.nlm.nih.gov/9241706/
22. Hwang S, Kim do Y, Kim M, et al. 2010. [Deficiencies in proteins C and S in a patient with idiopathic portal hypertension accompanied by portal vein thrombosis] Korean J Hepatol. 16: 176-181. Ref.: shorturl.at/ouBP5
23. Das SK, Ray A, Jana Ck, et al. 2011. Chronic portal vein thrombosis due to combined deficiency of protein C and protein S. J Indian Med Assoc. 109: 753-754. Ref.: shorturl.at/lJ023
24. Johnson NV, Khor B, Van Cott EM. 2012. Advances in laboratory testing for thrombophilia. Am J Hematol. 87: 108-112. Ref.: shorturl.at/fnvO0
25. Malkowski P, Pawlak J, Michalowicz B, et al. 2003. Thrombolytic treatment of portal thrombosis. Hepatogastroenterology. 50: 2098-2100. Ref.: shorturl.at/efIT4
26. Kim WS, Lee JW, Lee ES, et al. 2002. A case of isolated superior mesenteric vein thrombosis in acute pancreatitis. Korean J Gastroenterol. 40: 68-71. Ref.: shorturl.at/qwH36
27. Ferguson JL, Hennion DR. 2008. Portal vein thrombosis: an unexpected finding in a 28-year-old male with abdominal pain. J Am Board Fam Med. 21: 237-243. Ref.: shorturl.at/ayERT
28. Webster GJ, Burroughs AK, Riordan SM. 2005. Review article: portal vein thrombosis - new insights into aetiology and management. Aliment Pharmacol Ther. 21: 1-9. Ref.: shorturl.at/uwCKR
29. Friedrich-Rust M, Ong MF, Martens S, et al. 2008. Performance of transient elastography for the staging of liver fibrosis: a meta-analysis. Gastroenterology. 134: 960-974. Ref.: shorturl.at/quxR2
30. Sogaard KK, Astrup LB, Vilstrup H, et al. 2007. Portal vein thrombosis; risk factors, clinical presentation and treatment. BMC Gastroenterol. 7: 34. Ref.: shorturl.at/dnyAK
31. DeLeve LD, Valla DC, Garcia-Tsao G. 2009. Vascular disorders of the liver. Hepatology. 49: 1729-1764. Ref.:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6697263/
32. Turnes J, García-Pagán JC, González M, et al. 2008. Portal hypertension-related complications after acute portal vein thrombosis: impact of early anticoagulation. Clin Gastroenterol Hepatol. 6: 1412-1417. Ref.: shorturl.at/imHKQ
33. Ponziani FR, Zocco MA, Campanale C, et al. 2010. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol. 16: 143-155. Ref.: shorturl.at/prCHQ
34. Hall TC, Garcea G, Metcalfe M, et al. 2011. Management of acute non-cirrhotic and non-malignant portal vein thrombosis: a systematic review. World J Surg. 35: 2510-2520. Ref.: shorturl.at/afFSV
35. Amitrano L, Guardascione MA, Scaglione M, et al. 2007. Prognostic factors in noncirrhotic patients with splanchnic vein thromboses. Am J Gastroenterol. 102: 2464-2470. Ref.: shorturl.at/fmpJY


















