
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Case ReportDOI Number : 10.36811/jcri.2020.110015Article Views : 7Article Downloads : 5
Supravalvulaire Aortic Stenosis: Case Report
Redha Lakehal*, Soumaya Bendjaballah, Khaled Khacha, Baya Aziza, and Abdelmallek Brahami
Department of heart surgery, Ehs Dr Djaghri Mokhtar, Constantine, Algeria, Faculty of Medicine Constantine 03 Constantine / Algeria
*Corresponding Author: Dr. Lakehal Redha. Department of heart surgery, Ehs Dr Djaghri Mokhtar, Constantine, Algeria, Email: lakehal.redha@gmail.com
Article Information
Aritcle Type: Case Report
Citation: Redha Lakehal, Soumaya Bendjaballah, Khaled Khacha, et al. 2020. Supravalvulaire Aortic Stenosis: Case Report. J Case Rept Img. 2: 07-11.
Copyright: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2020; Redha Lakehal
Publication history:
Received date: 09 March, 2020Accepted date: 16 March, 2020
Published date: 17 March, 2020
Abstract
Introduction: Exceptional congenital heart disease (1 for 26000 birth) characterized by rétrécissement of aortic light. It can be isolated or part of William syndrome. The diagnosis is based on echocardiography. The intervention consists of an aortic root enlargement with Dacron patch. Surgery was indicated if gradient aortic left ventricle Superior of 50 mm hg. This clinical case is for us an opportunity to recall of this type of congenital aortic retrécissement.
Methods: We reported the observation of patient 17-year-old without history presented since one-year dyspnea on exersion, palpitation and syncopes. Physical examination: murmur systolic in aortic home without other abnormalities. Chest X ray: CTI: 0.48., ECG: RSR with HVL. Echocardiography: supravalvulaire aortic stenosis; mean gradient AO-LV: 46 mm hg, LV -aortic; LV 48/26 mm + HLV, RV: 20 mm. Exploration per-opératoire: hipoplasie of the left coronary sinus, anomaly of implantation of antero -external pillar of mitral valve, aortic bicuspidie type 1, absence of coronary anomalies. It has benefit under cardio-pulmonary bypass an enlargement of the left coronary sinus according to DOTY technique with Dacron patch and conservation of aortic valve.
Results: The immediate post-operative suites were favoured with gradient aortic –left ventricle drop to 20 mm hg. Conclusion: This is very rare congenital heart disease; echocardiography remains the key of diagnosis. It must be operated early. The prognostic is enhanced by the advances in surgical techniques. The treatment consists of surgery.
Keywords: Supravalvulaire aortic stenosis; Surgery; Cardiopulmonary bypass
Introduction
Supravalvular aortic stenosis (SVAS) is a heart defect that develops before birth. Perivalvar aortic stenosis (SVAS) is a fixed form of congenital left ventricular outflow tract (LVOT) obstruction that occurs as a localized or diffuse narrowing of the ascending aorta beyond the superior margin of the sinuses of Valsalva. It accounts for less than 7% of all fixed forms of congenital LVOT obstructive lesions. It is an exceptional congenital heart disease (1 for 26000 birth) characterized by stenosis of aortic light. It can be isolated or part of William syndrome. The precise etiology of SVAS is unknown [1-5]. The diagnosis is based on echocardiography. SVAS can be detected prenatally, particularly in patients with Williams syndrome, if it is revealed with fetal echocardiography. Cardiac catheterization or MRI may be indicated to evaluate the coronary artery or aortic arch anatomy [1-5]. Surgery is the primary treatment for SVAS. The choice of procedure varies with the type and severity of the stenosis [1-5]. The intervention consists of an aortic root enlargement with Dacron patch. Surgery was indicated if gradient aortic left ventricle Superior of 50 mm hg. If SVAS is not treated, the aortic narrowing can lead to shortness of breath, chest pain, and ultimately heart failure [1-5]. The severity of SVAS varies considerably, even among family members. Some affected individuals die in infancy, while others never experience symptoms of the disorder. This clinical case is for us an opportunity to recall of this type of congenital aortic stenosis [1-5].
Case Report
We reported the observation of patient 17-year-old without history presented since one-year dyspnea on exersion, palpitation and syncopes.
Physical examination: Murmur systolic in aortic focus without other abnormalities.
Chest X ray: Cardiothoracic index: 0, 48.
Figure 1: Chest X ray.
Electrocardiogram showed:
*Regular sinus rhythm with hypertrophy of left ventricular (HLV).
*Heart rate 80 battements / Mn.
Echocardiography showed:
*Supravalvulaire aortic stenosis,
* Aortic bicuspidie class 1;
*Mean gradient AO-LV: 46 mm hg;
*Max gradient AO-LV: 86 mm hg;
*Aortic ring 21mm, diameter sinus of Valsalva: 23mm, diameter of ascending aorta: 36mm.
*Left ventricular 45/26 mm + HLV,
*Right ventricular: 20 mm.
*Ejection fraction: 70%.
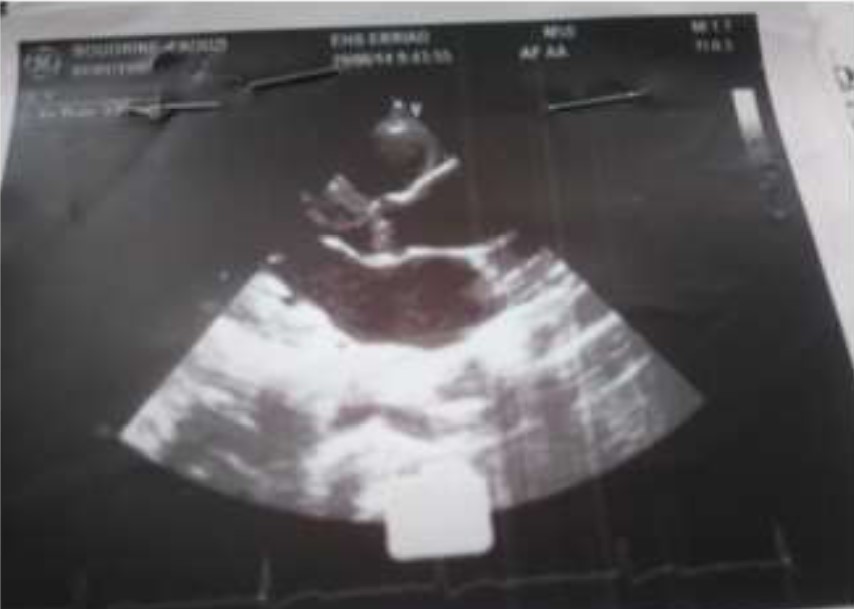
Figure 2: Transthoracic echocardiography.
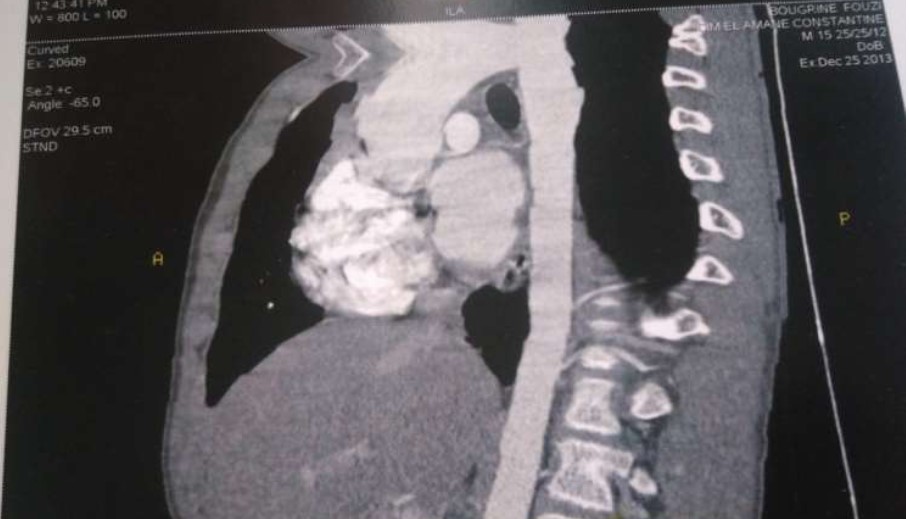
Figure 3: Transthoracic echocardiography.
Angiochest CT scan showed:
*Supravalvulaire aortic stenosis.
*Moderate dilatation of ascending aorta.
*Moderate HLV.
*Absence of coarctation.
Figure 4: Angiochest CT scan.
Surgery
The patient was opened under cardiopulmonary bypass established between the ascending aorta and right atrium. The approach was sternotomy. Operative exploration revealed: hipoplasie of the non-coronary sinus and right coronary sinus, anomaly of implantation of antero -external pillar of mitral valve, aortic bicuspidie class 1 and absence of coronary anomalies. The guesture was consisted enlargement of the non-coronary sinus and right coronary sinus according to the technique of Doty by Dacron patch and conservation of the aortic valve.
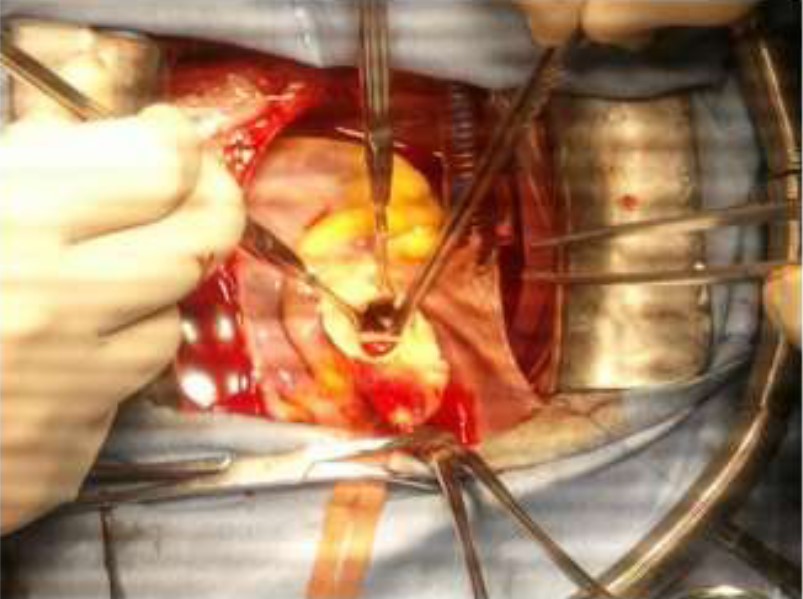
Figure 5: Operative view.
Results
* Duration of cardiopulmonary bypass: 133mn, aortic clamping: 92mn, Circulatory support: 38mn.
*Stay in intensive care unit: 48 hours.
*Intubation procedure: 14 hours.
*Post-operative stay: 07days.
*The immediate postoperative course was favoured with mean gradient aortic –left ventricle drops to 20 mm, good mobility of valves without reflux and conservation of ejection fraction of the left ventricle at 58%.
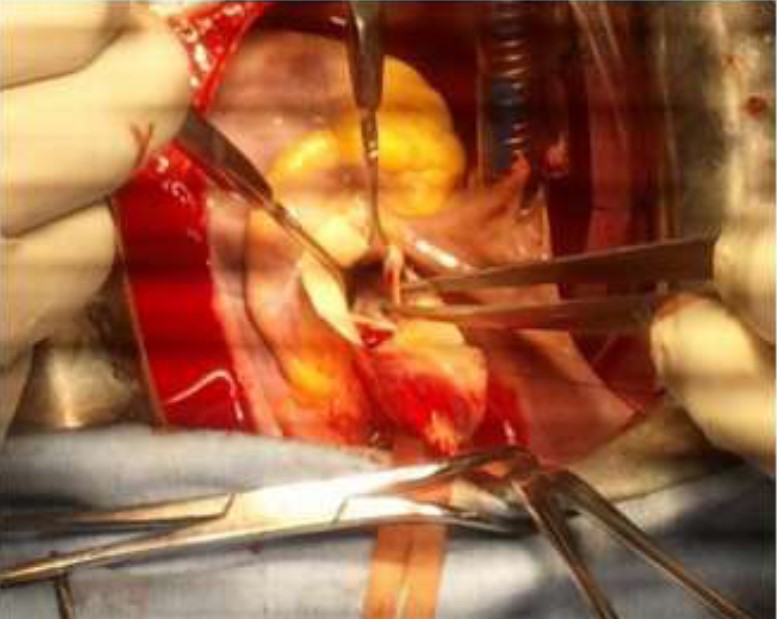
Figure 6: Operative view showing the aortic root with autotomy extended deep into the right and non-coronary sinuses.
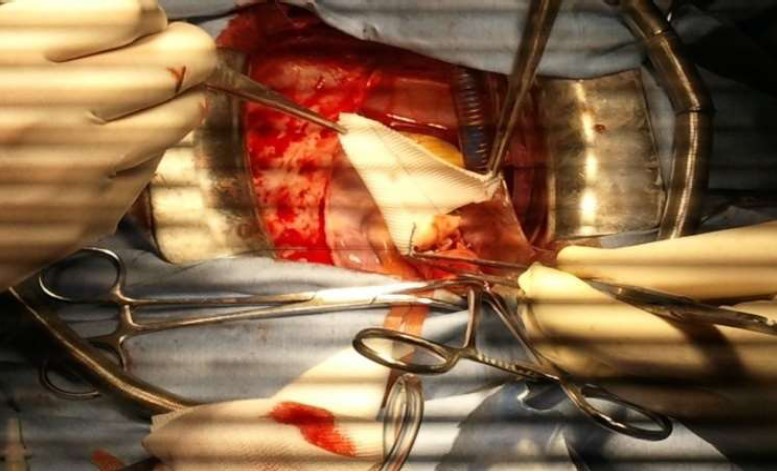
Figure 7: Operative view showing the accroissment of the aortic root with a Y Dacron patch.
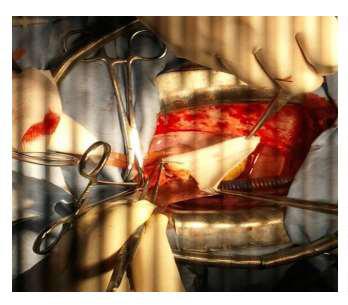
Figure 8: Operative view showing the accroissment of the aortic root with a Y Dacron patch.
Discussion
Surgical techniques for repair of supravalvular aortic stenosis (SVAS) include McGoon's one-patch , Doty's two-patch, and Brom's three-patch method [6] .Despite a longer cross clamp time, SVAS repair by Doty otoplasty restores normal hemodynamics and reduces the need for reoperation when compared with the classic one-patch technique [6-8]. The results from our work confirm that Doty’s technique for SVAS repair can be used routinely. It produced good early postoperative results and lower transaortic peak blood pressure gradients than those reported previously. The improvement of anatomic repair enables the expectation of a better long-term outcome than do other methods of surgical correction. Prognosis is influenced by the presence of genetic disorders, coronary artery lesions, and associated obstructive lesions of pulmonary arteries [9]. SVAS is a progressive lesion, whereas peripheral pulmonary artery stenosis remains unchanged or decreases in severity over time. The mortality rate is higher in patients with diffuse SVAS than in those with the localized form [11]. The risk of sudden cardiac death, including in patients who have undergone surgery, is 1 case per 1000 patient years and is 25-100 times higher than in the normal population [10-14]. Patients with SVAS are vulnerable to cardiac arrest or significant hemodynamic instability during induction of anesthesia secondary to hypotension and decreased coronary artery perfusion. Anatomic abnormalities that predispose individuals with SVAS and Williams syndrome to sudden death include coronary artery stenosis and severe biventricular outflow tract obstruction. The mechanisms for sudden death for both anatomic subgroups are believed to include myocardial ischemia, decreased cardiac output, and arrhythmia.
Conclusion
This is very rare congenital heart disease. Echocardiography remains the key of diagnosis. It must be operated early. The prognostic is enhanced by the advances in surgical techniques. The treatment consists of surgery.
Consent
The author obtained written, informed consent from the patient for the publication of this article.
References
1. Sharma BK, Fujiwara H, Hallman GL, et al. 1991. Supravalvar aortic stenosis: a 29-year review of surgical experience. Ann Thorac Surg. 51: 1031-1039. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/2039306
2. Stamm C, Friehs I, Yen Ho S, et al. 2001. Congenital supravalvar aortic stenosis: a simple lesion? Eur J Cardiothorac. 19: 195-202. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/11167112
3. Keating MT. 1995. Genetic approaches to cardiovascular disease. Supravalvular aortic stenosis, Williams syndrome, and long-QT syndrome. Circulation. 92: 142-147. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/7788908
4. Mencarelli L. 1930. Stenosi sopravalvolare aortica and anello. Arch Ital Istol Pat. 1: 829-841.
5. Metcalfe K, Rucka AK, Smoot L, et al. 2000. Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet. 8: 955-963. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/11175284
6. Stamm C, Friehs I, Ho SY, et al. 2001. Congenital supravalvar aortic stenosis: a simple lesion? Eur J Cardiothorac Surg. 19: 195-202. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/11167112
7. Merla G, Brunetti-Pierri N, Micale L, et al. 2010. Copy number variants at Williams-Beuren syndrome 7q11.23 region. Hum Genet. 128: 3-26. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/20437059
8. Pober BR, Johnson M, Urban Z. 2008. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Invest. 118: 1606-1615. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/18452001
9. Eisenberg R, Young D, Jacobson B, et al. 1964. Familial supravalvular aortic stenosis. Am J Dis Child. 108: 341-347. Ref.: https://bit.ly/2TK4myl
10. Chiarella F, Bricarelli FD, Lupi G, et al. 1989. Familial supravalvular aortic stenosis: a genetic study. J Med Genet. 26: 86-92. Ref.: https://bit.ly/2w2ZDPk
11. Ewart AK, Morris CA, Ensing GJ, et al. 1993. A human vascular disorder, supravalvular aortic stenosis, maps to chromosome 7. Proc Natl Acad Sci USA. 90: 3226-3230. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/8475063
12. Olson TM, Michels VV, Lindor NM, et al. 1993. Autosomal dominant supravalvular aortic stenosis: localization to chromosome 7. Hum Mol Genet. 2: 869-873. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/8364568
13. Frangiskakis JM, Ewart AK, Morris CA, et al. 1996. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell. 86: 59-69. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/8689688
14. Ewart AK, Jin W, Atkinson D, et al. 1994. Supravalvular aortic stenosis associated with a deletion disrupting the elastin gene. J Clin Invest. 93:1071-1077. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/8132745
15. Delius RE, Steinberg JB, L’Ecuyer T, et al. 1995. Long-term follow-up of extended aortoplasty for supravalvular aortic stenosis. J Thorac Cardiovasc Surg. 109: 155-162. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/advanced


















