
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Case ReportDOI Number : 10.36811/ojor.2019.110001Article Views : 2471Article Downloads : 30
Brachial plexus schwannoma in early adolescence. Case report
Pedro Clarós1*, Agnieszka Remjasz1-3 and Andrés Clarós1
1Clarós Clinic, Barcelona, Spain
2Department of Otorhinolaryngology at Stefan Zeromski Specialist Hospital, Cracow, Poland
3Scholarship in Clarós Clinic, Barcelona, Spain
*Corresponding author: Pedro Clarós, Clarós Clinic, Barcelona, Clarós Clinic, c./Vergós 31, 08017 Barcelona, Spain, Tel: +34932031212; +34639339252; Email: clinica@clinicaclaros.com
Article Information
Aritcle Type: Case Report
Citation: Pedro C, Agnieszka R, Andrés C. 2019. Brachial plexus schwannoma in early adolescence. Case report. Open J Otolaryngol Rhinol. 1: 01-07.
Copyright:This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2019; Pedro
Publication history:
Received date: 19 December, 2018Accepted date: 10 January, 2019
Published date: 11 January, 2019
Abstract:
Introduction: Primary tumors arising from the peripheral nerves are a rare occurrence. In 90% they are benign, although in a small percentage there is a risk of neoplastic transformation. Schwannomas and neurofibromas are the leading etiologies. They are commonly found in the neck and large nerves of limbs and arms, with a preference for regions of the elbow, wrist, and knee. The most vulnerable group of patients are people aged 20 to 50 years. The occurrence of this type of tumors in children is sporadic, especially if the tumor includes the brachial plexus.
Materials and Methods: The clinical case of a neurinoma in a supraclavicular region, affecting a 12-year-old girl. After a detailed diagnosis, the tumor finally came to be a schwannoma of a brachial plexus.
Discussion: Brachial plexus schwannoma is a rare find, although it should be considered in the differential diagnosis. A detailed search for signs of neurofibromatosis (von Recklinghausen's disease) should also be performed. Initial diagnostic imaging of cervical mass includes mainly MRI, thanks to which the surgeon can suspend the tumor of nervous origin, assess the vicinity of important nerves, vessels, and cervical spinal cord, as well as qualify for the surgery. Surgical treatment should be carried out after assessing the benefits and risks associated with the procedure, including irreversible damage to the brachial plexus. To maintain safety during the operation, the use of microsurgical instruments, microscopes or endoscopes should be considered by an experienced surgeon. Preserving nerve continuity is the essential goal of the treatment, especially in young, and active patients. Finally, the long-term observation is necessary for the assessment of potential tumor neoplastic transformation, nerve function of the brachial plexus and possible symptoms of neurofibromatosis.
Keywords: Brachial plexus tumor; Schwannoma in early adolescence; Neurofibromatosis; Supraclavicular swelling
Introduction
Primary tumors of brachial plexus are uncommon and comprise of about 5% neoplasm of the upper limbs [1-3]. They are dominated by neurofibromas and schwannomas, which are peripheral nerve tumors arising from the epineural sheath [4]. They were first called neurilemomas by Stout in 1935. Ehrlich and Martin introduced the name “schwannoma” in 1943. Neurinoma is another synonym of the schwannoma, presenting two types of tissues: an Antoni A type with areas of nuclear palisading (Verocay bodies) and the Antoni type B which is less cellular, more myxoid [1,3].
This group may be distinguished into benign tumors including traumatic neuromas, neurofibromas, and schwannomas or neurilemmomas, and a so-called peripheral nerve sheath malignant tumors [3]. Schwannomas and neurofibromas are the leading etiologies. They are very similar clinically, and only pathology diagnosis can discriminate them with certainty. A malignant transformation is a rare event [1]. In over 50% of cases, they occur as part of an NF1, but less than 5% of subjects with NF1 develop malignant transformation [5].
Schwannomas are peripheral nerves tumors, usually arising from C5 to C6 roots or upper trunk, very rarely from the lower trunk or lateral and medial cords [6-8]. They are also more peripheral and encapsulated in comparison to neurofibromas. Some studies have reported recurrences of schwannomas, the involvement of the same nerve by multiple schwannomas or the occurrence of two schwannomas within a few years of removing the first tumor [3,9].
This kind of neoplasm requires special attention due to diagnostic and surgical difficulties, which in particular applies to an unusual structure that can imitate malicious neoplasm, or large tumors adjacent to critical vessels and nerves. This unspecific presentation can lead to diagnostic dilemmas. We report a case of a 12-year-old active, practicing tennis girl with a right supraclavicular mass, which finally came to be a schwannoma of brachial plexus.
Case Report
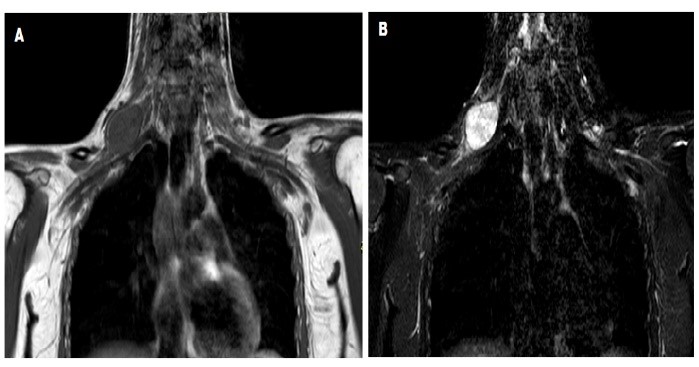
A 12-year-old girl with a past medical history of recurrent tonsillitis, consulted for a right lower cervical mass progressively enlarging for two years, as a first clinical manifestation. The patient also manifested some difficulties while playing tennis, and complained about a mild weakness of the right shoulder. She has no contributive personal or familial history. During an examination, a non-tender, isolated, round mass, measuring 4 cm in diameter, with a firm consistency was found in a supraclavicular region. It was mobile on both superficial and deep planes. Any neurological or sensory disorders were absent. The rest of the general examination was without any deviations from the norm. The diagnostic hypotheses were a congenital cyst, neurofibromatosis, metastatic adenopathy or apical lung tumor [10]. The patient underwent high-resolution real-time ultrasonography (US), computed tomography (CT) and magnetic resonance imaging (MRI). In ultrasonography, the tumor was presented as a well-defined hypoechoic, lobulated nodule, with posterior acoustic enhancement, and hypervascularization on a color Doppler ultrasonography. The CT scan of the neck revealed a 33 mm right supraclavicular nodular mass with heterogenic content showing small hypodense zones. It concluded on the diagnosis of possible metastatic adenopathy. The MRI showed an oval mass with a homogeneous signal on T1 and a higher heterogeneous signal on T2 acquisitions. The mass was highly contrasting and closely related to the right brachial plexus from which it seems to emerge. The MRI diagnosis was a tumor of a peripheral nerve (neurinoma) (Figure 1).
In order to deepen the diagnosis, the endoscopic exploration of the pharynx under general anesthesia was done. The investigation of the nasopharynx, oropharynx, and hypopharynx were made towards the presence of atypical neoplastic changes, and to perform a nasopharyngeal biopsy. During the same operation, a fine needle aspiration of the cervical mass was performed. The result of the nasopharynx sample was a moderate unspecific lymphoid hyperplasia of the mucosa, without atypia or malignancy. The fine needle aspiration was muco-hemorrhagic, without evidence of other significant cellular elements.
Figure 1: The comparison of MRI in T1-weighted scans of the neck and thorax mass before contrast (A) and after contrast enhancement (B). The tumor lies between the scalene muscles and the pectoralis minor muscle, just a few millimeters from the pleura.
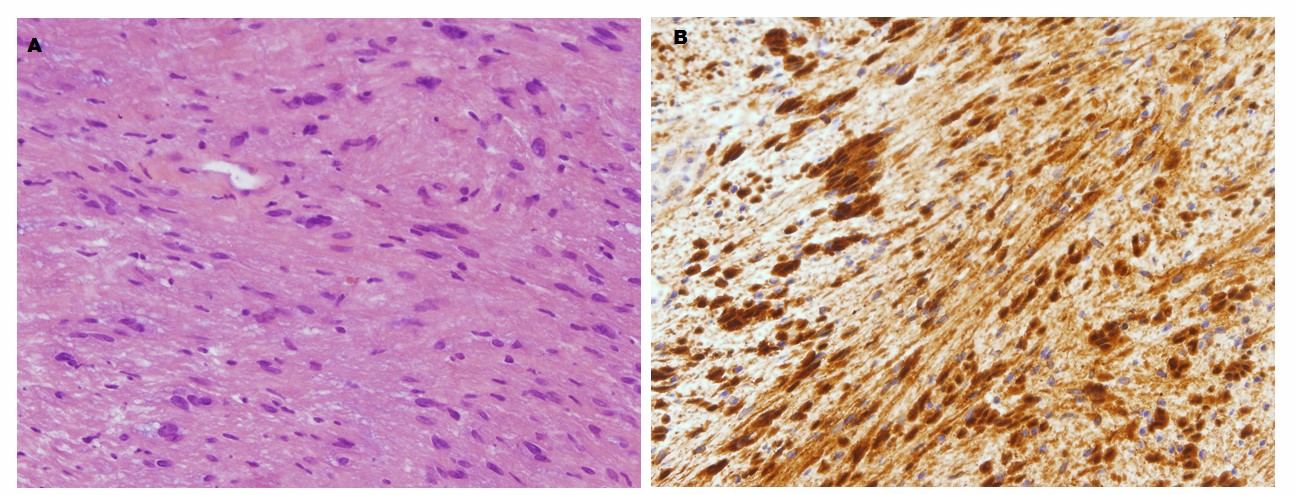
The surgical procedure was carried out under general anesthesia, by an anterior approach. The linear incision was done above the mass in a cervical crease. The hard whitish mass was found deep in the neck, with a stalk emerging from a cervical root of the brachial plexus (Figure 2). The blood supply of the tumor mass came from the subclavian-axillary trunk. The tumor was resected using the nerve stimulator and microsurgical technique, in the company of a microscope. After the excision of the tumor, the material was sent for histopathological examination. The histopathology analysis and immunohistochemical study (S-100 protein and Ki-67) confirmed the diagnosis of schwannoma. (Figure 3).
Postoperatively, the patient complained of a pain irradiating from the right axilla to the ulnar region that subsided after a week. After confirmation of the diagnosis of schwannoma, we surveyed family members and a genetic test that proved non-contributory to the determination of neurofibromatosis type 1 or 2 (NF1 or NF2). After nine years of follow-up, no paresthesia or tumor recurrence has been evident up today. The patient feels good, negates any pain or weakness of muscle strength, and continues to play tennis regularly.
Figure 2: Intraoperative view of the tumor in the right supraclavicular region (A), and a macroscopic view of its cross section (B). The mass is heterogeneous.
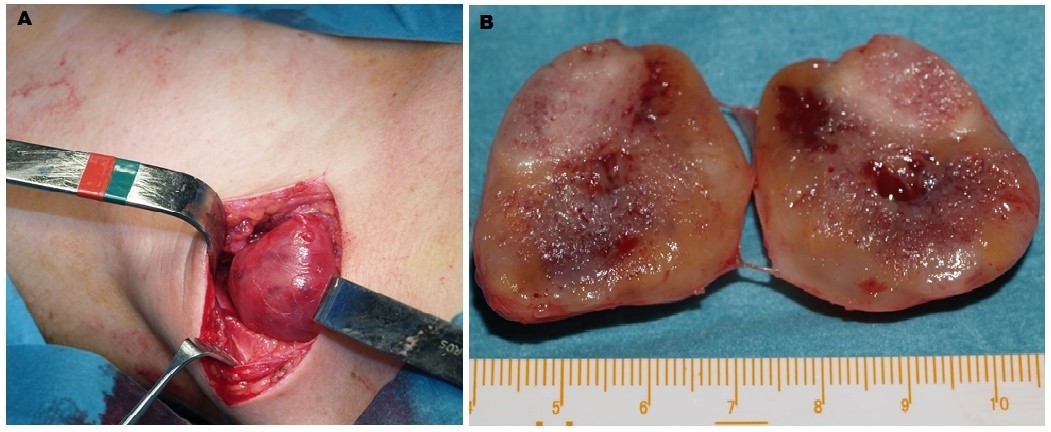
Figure 3: Photomicrographs of the slides. A. Spindle cells arranged in an irregular pattern, with a slightly eosinophilic cytoplasm and partially elongated nuclei (x20). B. Strong cell immunoreactivity to S-100 protein characteristic for Schwann cells (x20).
Discussion
Primary tumors arising from peripheral nerves characterized by a rare prevalence [1-3] and occurrence on every stage of life, but predominantly between 20 and 50 years of age [10]. As in a study conducted by Maiuri et al. there were four patients from 18 to 32 years of age with schwannomas of the brachial plexus presented [8]. Our case is the exception from the rule due to the early onset and absence of a family history of neurofibromatosis or personal history of any other tumors of peripheral nerve origin. Regarding Kehoe et al. study, only two cases of schwannomas under the age of 18 years were presented [3].
Clinically, there is no pathognomonic sign or symptom of those tumors. Schwannomas can either be solid or cystic [2]. According to Kehoe et al., before a tumor of a limb, an anatomical consideration such as mobility in the transversal axis, and tethering in the longitudinal axis of the limb can only lead to the conclusion of a peripheral nerve tumor [3]. However, this rule of thumb is neither universal since in the case reported; the tumor was mobile in both planes. Regarding Maiuri et al., all four schwannomas were hypodense on plain CT and moderately hyperdense after contrast administration, which may help to distinguish these tumors from neurofibromas [8]. On the other hand, Jee et al., in 2004 found it difficult to differentiate schwannomas and neurofibromas using MRI scans [11]. In some cases, the high-resolution and color Doppler ultrasonography may be useful in differential diagnosis, showing a nerve eccentrically entering the mass of the schwannoma, which in turn is not seen in the case of neurofibroma [12]. In our case, the hard consistency of the tumor and its site suggested malignant adenopathy that the CT-scan also hypothesized. This could be ruled out by the biopsy of the cavum, the fine needle aspiration of the mass and the MRI scans which could predict the final diagnosis. In case series of peripheral nerve tumors, published in 1995 by Kehoe et al., an accurate preoperative diagnosis could be made only in 42% of patients [3]. They explained that low rate by the fact that at this time, modern imaging techniques such as MRI were not developed. The latter is more helpful for differentiating neurofibromas and malignant peripheral nerve sheath tumors according to Bhargava et al., thanks to the “target sign” [13]. Information provided by CT or MRI may be useful to estimate the relationship to the neural and vascular structures which is necessary before planning the surgery [14,15]. However, due to the small size of neural trunks or cords, especially of the brachial plexus, it is difficult to identify the position and relation to other structures in comparison to larger nerves of the limbs correctly [8]. It should be kept in mind as a differential diagnosis of a cervical MRI contrast-enhancing well-defined mass [16,17].
Pathology remains the only way to obtain a particular diagnosis, by Hematoxylin-eosin stain, immunostaining for S-100 or Sox10 protein. Despite a well-characterized lack of speci?city, some pathologists still routinely employ S100 in the diagnosis of neural crest-derived tumors. In 1982, Nakajima et al. [18] first described the results of S100 protein staining and its variably expressed in tissues of adipocytic, chondroid or myoepithelial derivation. Other studies described that S100 was also expressed in various non-neural crest-derived tumors including synovial sarcoma, rhabdomyosarcoma or Ewing sarcoma [19-21]. In contrast, Sox10 is recently named as neural crest stem cell marker and is more sensitive and specific marker than S100, especially for schwannoma and melanocytic tumors [22,23]. Intact Sox10 signaling is vital for normal central and peripheral nervous system myelination [24]. However, in some cases schwannoma can demonstrate morphologic similarities to ?brosarcoma, leiomyosarcoma or even synovial sarcoma which manifests itself in the absence of Sox10 expression. Distinguishing between these entities by morphology can be particularly challenging on small needle biopsy specimens. In such cases, additional immunohistochemical staining with TLE1 may be necessary for differential diagnosis. Also, a Ki-67 stain showing a low proliferative index indicates a benign tumor. Some authors state that both Sox10 and S100 should be used for diagnostic purposes in the realm of soft-tissue neoplasms [25]. Various studies proved that schwannoma-like regions displayed strong S100 staining, in contrast to more different and limited S100 reactivity in neurofibromatosis areas [3]. Fine et al. [26] noticed that calretinin was detected in almost all schwannomas and only a small percentage of neurofibromas, suggesting it could be a reliable marker for differentiating schwannomas from neurofibromas. In another study, Gray et al. [27] proved that neurofibromas are characterized by high expression of XIIIa factor in contrast to schwannomas. Work on research on new factors facilitating differential diagnosis is still ongoing.
Regarding the young age of the patient, it is worth mentioning the genetic background of these types of tumors. Schwannomas occur both as a sporadic tumor and in the dominantly inherited familial cancer syndrome neurofibromatosis type 2 (NF2). The disease is caused by some mutations in the NF2 gene in the 22q12.2 locus coding for the merlin protein [28]. The NF2 gene has a smaller length than NF1; consists of 16 constitutive and one alternatively excised exon. Alternative splicing results in at least two forms of protein with different tissue expression. The function of a protein is to mediate interactions between the external environment of the cell and the cytoskeleton. Recent studies have also presented that there is a rearrangement of HTRA1 and SH3PXD2A genes in the subgroup of schwannoma tumors. In normal cells, these genes are physically separated, but in schwannomas, they form the fusion genes. The fusion gene has a cellular function of promoting tumor development, suggesting that it acts as a "driver" of carcinogenesis. Since the fusion is unique to cancer cells, it is the ideal candidate for targeted treatment because the abolition of its function would represent a tumor-specific approach that would save normal cells in the body. Further research aims to focus on the functioning of the fusion gene for diagnostic and therapeutic development [29].
Treatment of this kind of tumors is by surgical excision. Radiotherapy is inadvisable and can lead to neoplastic transformation [30]. The operation should be decided after balancing the symptoms and the risks to the plexus. Complete surgical removal of brachial plexus schwannomas should be achieved due to the high risk of recurrence if the small fragments of the tumor are left. In the cases of large tumors, the enucleation may be enough, while there is the low possibility of recurrence. Although, the excision of a peripheral nerve benign tumor may not be necessary if there are no other symptoms than the existence of the mass since it can be deleterious. Kehoe et al. had only 5% of preoperative neurologic deficits in their series and up to 11.5% of postoperative deficits being a pain, motor, sensory and mixed. In this perspective, excision of neurofibromas, which are more closely related to the nerve, is more likely to result in neurologic deficit [3]. In symptomatic cases, it is advisable to remove the tumor, and the results presented in studies of Maiuri et al. or Rashid et al. are either convincing, due to the fact that patients have no post-operative complications [8,31]. The microsurgical technique is indicated due to the different distribution of nerve fibers in the tumor. This allows for complete excision of the tumor and the preservation of as many nerve fibers as possible, which is important in maintaining the motor function of the patient.
Postoperative results are generally satisfactory in patients who underwent the complete removal during the first procedure, with a low risk of motor dysfunction (4%) [8]. However, the situation is different when comparing to patients who need the secondary look. Then, the anatomical conditions and the proper differentiation of nerve structures might cause difficulties even for the well-experienced surgeon. It is worth mentioning that, the long-term observation is necessary for the assessment of potential tumor neoplastic transformation, nerve function of the brachial plexus and possible symptoms of neurofibromatosis.
Conclusion
Schwannomas are rare in peripheral nerves and unique in children without any past familial history of neurofibromatosis. However, it should be kept in mind as a differential diagnosis of a cervical MRI contrast-enhancing well-defined mass. Due to the young age of the patient, the surgeon should always look for other symptoms characteristic for neurofibromatosis and consider performing genetic counseling. In order to avoid schwannomas recurrences or nerve damages, it is necessary to perform the tumor excision entirely using microsurgical tools. The surgical excision should be decided after balancing the symptoms and the risk to the plexus, including the patient’s age and preferences. The long-term follow-up should be obligatory due to the probability of tumor recurrence, neoplastic transformation, and the possibility of developing a set of symptoms - neurofibromatosis.
References
- Forte A, Gallinaro LS, Bertagni A, et al. 1999. Neurinomas of the brachial plexus: case report. Eur Rev Med Pharmacol Sci. 3: 19-21.[Ref.]
- Chen F, Miyahara R, Matsunaga Y, et al. 2008. Schwannoma of the brachial plexus presenting as an enlarging cystic mass: report of a case. Ann Thorac Cardiovasc Surg. 14: 311-313.[Ref.]
- Kehoe NJ, Reid RP, Semple JC. 1995. Solitary benign peripheral-nerve tumors. Review of 32 years' experience. J Bone Joint Surg Br. 77: 497-500.[Ref.]
- Ganju A, Roosen N, Kline DG, et al. 2001. Outcomes in a consecutive series of 111 surgically treated plexal tumors: a review of the experience at the Louisiana State University Health Sciences Center. J Neurosurg. 95: 51-60.[Ref.]
- Whitaker WG, Droulias C. 1976. Benign encapsulated neurilemmonas: a report of 76 cases. Am Surg. 42: 675-678.[Ref.]
- Lusk MD, Kline DG, Garcia CA. 1987. Tumors of the brachial plexus. Neurosurgery. 21: 439-453.[Ref.]
- Fisher RG, Tate HB. 1970. Isolated neurilemmomas of the brachial plexus. J Neurosurg. 32: 463-467.[Ref.]
- Maiuri F, Donzelli R, Benvenuti D, et al. 2001. Schwannomas of the Brachial Plexus - Diagnostic and Surgical Problems. Zentralbl Neurochir. 62: 93-97.[Ref.]
- Chen BH, Huang PW, Kao CC, et al. 2011. Recurrent Penile Schwannomas. J Med Sci. 31: 223-225.[Ref.]
- Horowitz J, Kline DJ, Keller SM. 1991. Schwannoma of the brachial plexus mimicking an apical lung tumor. Ann Thorac Surg. 52: 555-556.[Ref.]
- Jee WH, Oh SN, McCauley T, et al. 2004. Extra axial neurofibromas versus neurilemmomas: discrimination with MRI. Am J Roentgenol. 183: 629-633.[Ref.]
- Tsai WC, Chiou HJ, Chou YH, et al. 2008. Differentiation between schwannomas and neurofibromas in the extremities and superficial body: the role of high-resolution and color Doppler ultrasonography. J Ultrasound Med. 27: 161-166.[Ref.]
- Bhargava R, Parham DM, Lasater OE, et al. 1997. MR imaging differentiation of benign and malignant peripheral nerve sheath tumors: use of the target sign. Pediatr Radiol. 27: 124-129.[Ref.]
- Catagno AA, Shuman WP. 1987. MR imaging in clinically suspected brachial plexus tumor. Am J Roentgenol. 149:1219-1222.[Ref.]
- Kline DG, Judice DJ. 1983. Operative management of selected brachial plexus lesions. J Neurosurg. 58: 631-649.[Ref.]
- Herms TE, Burge PD, Wilson DJ. 1997. The role of magnetic resonance imaging in the management of peripheral nerve tumors. J Hand Surg Br. 22: 57-60.[Ref.]
- Kneelands JB, Kellman GM, Middleton WD, et al. 1987. Diagnosis of disease of the supraclavicular region by use of MR imaging. Am J Roentgenol. 148: 1149-1151.[Ref.]
- Nakajima T, Watanabe S, Sato Y, et al. 1982. S-100 protein in Langerhans cells, interdigitating reticulum cells and histiocytosis X cells. Gann. 73: 429-432.[Ref.]
- Shimada H, Newton WA Jr, Soule EH, et al. 1988. Pathologic features of extraosseous Ewing's sarcoma: a report from the Intergroup Rhabdomyosarcoma Study. Hum Pathol. 19: 442-453.[Ref.]
- Coindre JM, de MA, Trojani M, et al. 1988. Immunohistochemical study of rhabdomyosarcoma. Unexpected staining with S100 protein and cytokeratin. J Pathol. 155: 127-132.[Ref.]
- Fisher C, Schofield JB. 1991. S-100 protein positive synovial sarcoma. Histopathology. 19: 375-377.[Ref.]
- Paratore C, Goerich DE, Suter U, et al. 2001. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development. 128: 3949-3961.[Ref.]
- Nonaka D, Chiriboga L, Rubin BP. 2008. Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol. 32: 1291-1298.[Ref.]
- Pingault V, Bondurand N, Le CC, et al. 2001. The SOX10 transcription factor: evaluation as a candidate gene for central and peripheral hereditary myelin disorders. J Neurol. 248: 496-499.[Ref.]
- Nonaka D, Chiriboga L, Rubin BP. 2008. Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol. 32: 1291-1298.[Ref.]
- Fine SW, McClain SA, Li M. 2004. Immunohistochemical Staining for Calretinin Is Useful for Differentiating Schwannomas From Neurofibromas. Am J Clin Pathol. 122: 552-559.[Ref.]
- Gray MH, Smoller BR, McNutt NS. 1990. Immunohistochemical Demonstration of Factor XIIIa Expression in NeurofibromasA Practical Means of Differentiating These Tumors From Neurotized Melanocytic Nevi and Schwannomas. Arch Dermatol. 126: 472-476.[Ref.]
- Hulsebos TJ, Plomp AS, Wolterman RA, et al. 2007. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 80: 805-810.[Ref.]
- Sameer A, Shahrzad J, Mark W, et al. 2016. The genomic landscape of schwannoma. Nat Gen.[Ref.]
- Kragh LV, Soule EH, Masson JK. 1960. Benign and malignant neurilemmomas of the head and peck. Surg Gynecol Obstet. 111: 211-218.[Ref.]
- Rashid M, Salahuddin O, Yousaf S, et al. 2013. Schwannoma of the brachial plexus; report of two cases involving the C7 root. Journal of Brachial Plexus and Peripheral Nerve Injury. 8: 12.[Ref.]


















