
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Mini ReviewDOI Number : 10.36811/ojpsr.2019.110006Article Views : 73Article Downloads : 40
Therapeutic targets to reduce the contribution of pulmonary neutrophilic inflammation towards obesity-associated co-morbidities: a mini-review
Claveles Casas FN1,2, Barrera FS2, Lopez CM1, Chacon I. del V2, Di Sciullo MP1, Ramirez DC1* and Gomez Mejiba SE2*
1Laboratory of Experimental & Translational Medicine. IMIBIO-SL, CONICET-National University of San Luis, San Luis, 5700 San Luis, Argentina
2Laboratory of Experimental Therapeutics & Nutrition. IMIBIO-SL, CONICET-National University of San Luis, San Luis, 5700 San Luis, Argentina
*Correspondig Author: Dr Dario C Ramirez and Dr Sandra E Gomez Mejiba. IMIBIO-SL, CONICET-National University of San Luis, San Luis, 5700 San Luis, Argentina, Emails: ramirezlabimibiosl@ymail.com; sandraegomezmejiba@yahoo.com
Article Information
Aritcle Type: Mini Review
Citation: Claveles Casas FN, Barrera FS, Lopez CM, et al. 2019. Therapeutic targets to reduce the contribution of pulmonary neutrophilic inflammation towards obesity-associated co-morbidities: a mini-review. Open J Pharm Sci Res. 1: 123-133.
Copyright: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2019; Claveles Casas FN
Publication history:
Received date: 01 August, 2019Accepted date: 08 August, 2019
Published date: 10 August, 2019
Abstract
Epidemiology and experimental models have shown a close link between adipose tissue inflammation, systemic inflammation and pulmonary neutrophilic inflammation, which predispose obese patients to pulmonary diseases, obesity-associated co-morbidities and cancer. Increased content and activation of neutrophils in the lung microvasculature, resulting from peripheral activation of neutrophils, and increased adhesion of neutrophils to the lung microvasculature are important factors explaining the increased susceptibility of obese patients towards respiratory diseases and loss of insulin sensitivity. Mechanism-based therapies to break this link are urgently needed to reduce pulmonary damage in obesity, due to the growing prevalence of obesity world-wide. Current research suggests that these approaches should be focused on, one or more of the following: reduction of macrophage activation at the adipose tissue, healthy growing of adipose tissue by induction of Nrf-2, inhibition of NF-?B activation, reduction of circulating neutrophil activation, blocking adhesins/selectins, inhibition of neutrophil activation by targeting NADPH oxidase-2 activation, inhibition of myeloperoxidase activity and scavenging of hypochlorous acid. These strategies are expected to reduce adipose tissue inflammation, peripheral inflammation, pulmonary neutrophilic inflammation and obesity-associated co-morbidities.
Keywords: Obesity; Adipose tissue inflammation; Systemic inflammation; Neutrophil, Myeloperoxidase; Therapeutic target
Adipose Tissue and Systemic Inflammation in Obesity
Obesity is a frequent, costly and serious chronic inflammatory disease. This disease is characterized by a state of low-grade chronic inflammation resulting of a chronic positive energy-balance [1,2]. When adipose tissue capacity to store energy is overwhelmed, excess free fatty acids (FFAs) increase in circulation [3] where it can activate a number of vasculatures and circulating innate immune cells, such as monocytes and neutrophils [4,5]. Increased circulating activated neutrophils have been observed in obese patients [6]. This activation process may be caused by inflammatory factors such as end-oxidation products of macromolecules, as well as pro-inflammatory cytokines released by the inflamed adipose tissue [7,8]. Intestinal microbiota is also a factor to consider due to the increased intestinal permeability to endotoxin that can translocate into the systemic circulation and activate peripheral cells, as well as distant tissue vasculatures [1-9].
Adipose tissue inflammation is a multifactorial process that involves a number of causes, including adipose extracellular matrix stress, endotoxin translocation from the gut into circulation, macrophage infiltration, hypoxia, endoplasmic reticulum-stress and mitochondrial dysfunction [2,7,10,12]. Products released by the inflamed adipose tissue reach systemic circulation, where they can activate leukocytes and other tissue cells.
Adipose tissue macrophages derive from circulating monocytes. Under normal conditions these monocytes are differentiated into macrophages with an M2 anti-inflammatory phenotype; and their function is the patrol of the tissue for damaged/death cells, tissue reparation and remodeling [10]. However, under metabolic stress conditions caused by hypoxia, endoplasmic reticulum (ER)-stress and FFAs, monocytes are recruited in large numbers and differentiated into pro-inflammatory M1-macrophages [10,13]. The pro-inflammatory phenotype of these macrophages is caused mainly by activation of the nuclear factor-?B (NF-?B) signaling pathway [14]. This activation leads to expression of a number of pro-inflammatory cytokines/chemokines and large production of reactive biochemical species. Adipocyte hypertrophy and monocyte infiltration are the most relevant events in adipose tissue inflammation, because they are associated with the phenotypic switch of macrophage towards M1-proinflammatory cells located at hypoxic areas within the adipose tissue, crown-like structures [11,13,15].
Deregulated secretion of inflammatory mediators by a growing mass of fat tissue in obesity is a risk factor for biochemical stress in the lung of obese patients [16]. Figure 1 shows some of the pulmonary complications observed in obese patients and in animal models of obesity. These complications are the result of mechanical and biochemical pressures in obese patients and animals. Adipokines are proteins that are mainly or exclusively produced by adipocytes, and responsible for many of the pulmonary complications in obesity [17]. Examples include leptin, resistin and adiponectin, which have potential immunomodulatory effects in pulmonary cells [17,18]. Genetically obese ob/ob mice, which produce a mutated, non-functional form of leptin, show many of the same inflammatory changes as other models of obesity (including diet-induced obese mice), and ob/ob mice become both insulin resistant and diabetic [19]. These results indicate that leptin may not have a particularly important role in obesity-induced inflammation [20]. Adiponectin is considered to be an anti-inflammatory, insulin-sensitizing and cardioprotective protein [17,21-23]. It may cause these effects in several pathways; for example, by inducing anti-inflammatory cytokines such as IL-10 and IL-1Ra, through vascular mechanisms includ-ing enhancement of nitric oxide bioavailability, or by reducing endothelial cell–leukocyte adhesion [24,25].
Figure 1: Pulmonary complications of obesity. Image shows a number of pulmonary complications observed in obese patients caused by adipose tissue-derived systemic inflammation. COPD, chronic-obstructive pulmonary disease.

In obesity, a low-grade chronic state of inflammation is responsible for multiple organs dysfunction [10]. Systemic inflammation produced by the inflamed fat has been correlated with the development of insulin resistance, a hallmark of type 2 diabetes mellitus (T2DM) as well as a number of chronic inflammatory diseases and cancer [2,26].
The Lung Microvasculature Homes More Neutrophils in Obesity
Inflammation mediators (pro-inflammatory cytokines/chemokines and macromolecule end-oxidation products) released by the adipose tissue [11] may activate both, circulating neutrophils and peripheral microvasculatures (Figure 2). Therapies aimed at reducing activation of neutrophils, recruitment of neutrophils into tissues and adhesion molecules in microvasculature may have clinical benefit in the future to overcome tissue damage induced by excessive leukocyte infiltration [6,27]. In addition, obese and leptin-supplemented mice are more sensitive to ozone-induced lung inflammation than their lean counterparts [28]. This evidence suggests that in obese patient’s leptin and/or resistin may cause lung inflammation by increasing neutrophil content, and possibly their activation, in the lung microvasculature [29-31]. In pathological conditions such as asthma and chronic obstructive pulmonary disease (COPD), it is likely that the tracheal-bronchial circulations are pivotal in the recruitment of circulating leukocytes [29,32]. Furthermore, the mechanism of the airway circulation for the recruitment of inflammatory cells in obesity after the inhalation of airborne pollutants requires further studies.
Figure 2: Obesity, pulmonary neutrophilic inflammation and obesity-associated metabolic abnormalities.
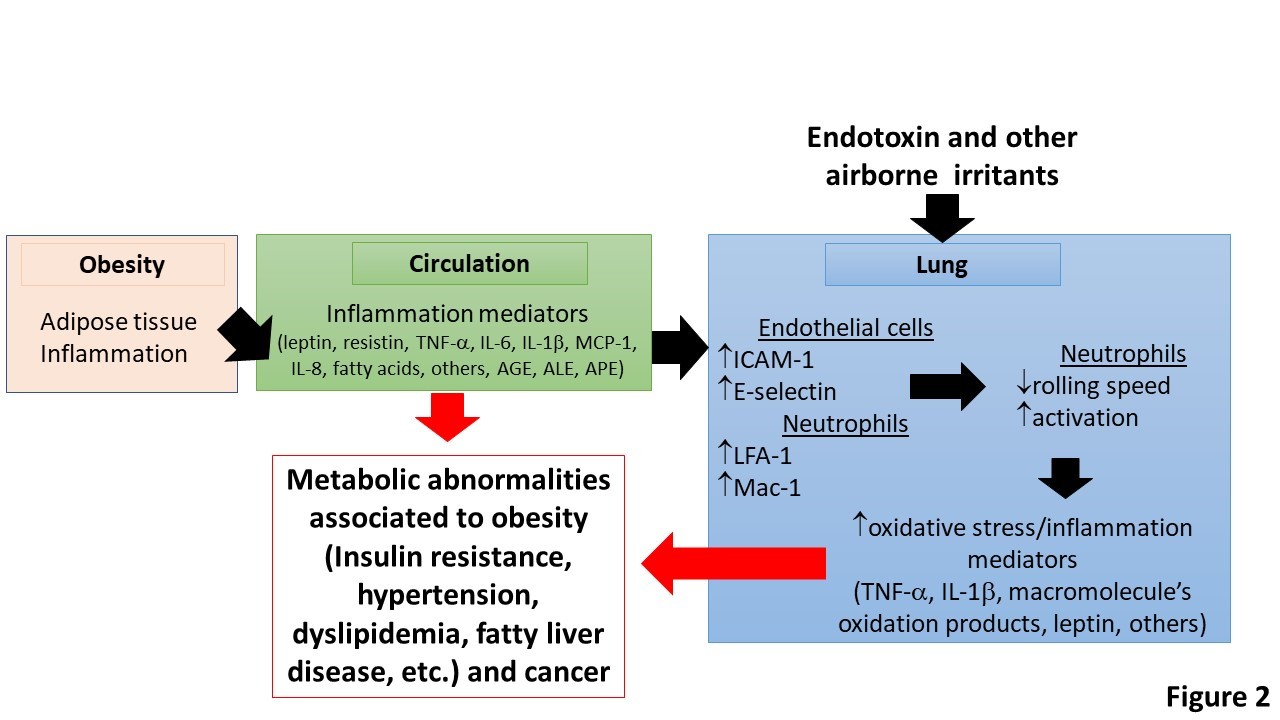
Adipose tissue inflammation in obesity produces a number of inflammation mediators. These cause activation of neutrophils in circulation and endothelial cells in the lung. This process results in increased content and activation of neutrophils and MPO-promoted oxidation reactions and pulmonary inflammation. Under this condition the lung acts as an extra source of inflammation mediators that contribute towards systemic inflammation and worsen obesity-associated co-morbidities and cancer. AGE, advanced-glycation end-products; ALE, Advanced-lipid peroxidation end-products; APE, Advanced-protein oxidation-end products.
Neutrophils are key sentinel cells of the innate immune system and are the premier cellular responders to acute inflammation [33,34]. For example, in models of acute lung injury ranging from acid aspiration to ischemia- reperfusion, depletion of neutrophils before the injury stimulus protects rabbits, rats and mice, respectively, from lung injury. These noninfectious models differ from the most common clinical causes of acute lung injury, pneumonia and sepsis, in which neutrophils are needed to control the infection. Nonetheless, animal work has demonstrated that the pro-inflammatory response of the neutrophil can lead to an increase in endothelial and epithelial permeability and, in the case of sepsis, shock and global organ injury [35,36].
The lung microcirculation has a concentration and traffic of neutrophils 30-100-times higher than any other large vasculatures in the human body, which might be further increased during obesity, making it a potential source of oxidants, oxidized products and pro-inflammatory cytokines upon exposure to air pollutants [37-40]. Airway inflammation requires an orchestrated series of molecular events whereby inflammatory cells leave the airway vasculature and migrate within the airway space [41]. Leukocytes are initially tethered to the endothelial cell surface of post-capillary venules, then roll, before firmly adhering and subsequently migrating out of the vasculature [42]. In obesity, circulating inflammation mediators derived from an inflamed adipose tissue, have been shown to produce oxidative stress in the brain and muscle tissue, both cardiac and skeletal, but the information in lung is limited [43]. Among the pro-inflammatory adipokines secreted in high quantities by the inflamed fat tissue, leptin, TNF-?, MCP-1, IL-1? and resistin have been shown to increase intercellular-adhesion molecule-1 (ICAM-1) expression in endothelial cells and to slow neutrophil rolling [16]. These adipokines and other inflammatory mediators in the serum of obese patients are thought to be a leading cause of systemic inflammation and thus obesity-associated metabolic disorders [37,40].
Leukocyte-endothelial cell interactions are mediated by various cell adhesion molecules [44]. These interactions are important for leukocyte extravasation and trafficking in all domestic animal species. An initial slowing of leukocytes on the vascular endothelium is mediated by selectins [45]. This event is followed by activation of ?2-integrins after leukocyte exposure to cytokines and pro-inflammatory mediators, adherence of leukocyte ?2 integrins to vascular endothelial ligands (e.g., ICAM-1), extravasation of leukocytes into tissues through tight junctions of endothelial cells mediated by platelet and endothelial cell adhesion molecule-1 (PECAM-1), and perivascular migration through the extracellular matrix via ?1 integrins [42,46]. Inhibiting excessive leukocyte egress and subsequent free radical-mediated damage caused by leukocyte activation may attenuate or eliminate tissue damage [47]. Several methods have been used to modify leukocyte infiltration in various animal models [48]. These methods include nonspecific inhibition of pro-inflammatory mediators and adhesion molecules by nonsteroidal anti-inflammatory drugs and glucocorticoids, inhibition of cytokines and cytokine receptors, and inhibition of specific types of cell adhesion molecules, with inhibitors such as peptides and antibodies to ?2 integrins, and inhibitors of selectins, ICAMs, and vascular cell adhesion molecule-1 (VCAM-1) [46]. By understanding the cellular and molecular events in leukocyte-endothelial cell interactions, therapeutic strategies are being developed in several animal models and diseases in which neutrophilic inflammation plays a role.
Oxidations Promoted by Pulmonary Neutrophilic Inflammation and Systemic Inflammation in Obesity
Previously an increased activation of neutrophils in the systemic inflammation induced by adipose tissue inflammation has been reported [49]. We have also shown increased of neutrophil accumulation and activation in the lung of diet-induced obesity mouse model [50]. Neutrophil activation was associated with more oxidative stress and inflammation in the lung and systemic environments [17,51,52]. This effect caused reduced insulin sensitivity that was improved by decreasing neutrophil homing, myeloperoxidase activation or by scavengers of HOCl (unpublished data). The main players in causing lung inflammation induced by environmental and metabolic stressors are neutrophils and myeloperoxidase (MPO), an enzyme contained in neutrophils’ azurophilic granules [51,53]. MPO is the only mammalian enzyme that, under physiological conditions, produces HOCl [54]. MPO can produce a number of hypohalous (e.g., HOCl, IOH and BrOH) and pseudo-hypohalous (e.g., SCNOH) compounds [55]. MPO uses H2O2 to oxidize chloride anions into hypochlorous acid/hypochlorite (HOCl/−OCl, from now on HOCl), a potent oxidizing agent [56]. Hypochlorous acid further reacts with other biological molecules to generate secondary reactive species, such as chloramines and NO2Cl [56,57]. Determination of blood MPO and HOCl-modified proteins have been suggested a predictive biomarker of cardiovascular accident [52].
Activation of neutrophils and production of HOCl in the lung vasculature causes oxidative stress and inflammation in the lung [50,52]. On the other hand, inhalation of air pollutants is associated with altered serum biomarkers of oxidative stress and inflammation (BOSI) and a higher incidence of insulin resistance in swine farm workers [58]. The incidence of cardiovascular diseases has been reported to increase in humans driving cloudy routes during rush hours [59,60]. The prevalence of airway sensitization in obese is higher than lean subjects [51,61] (See Figure 1). This evidence has been also supported in obese experimental animals [32]. Therefore, it is possible that inhalation of air pollutants increases the incidence of systemic inflammation and obesity-associated metabolic complications in obese patients (BMI>30 Kg/m2) in relation to normal weight subjects [2]. This disparity between obese and lean patients to develop obesity-associated diseases upon inhalation of air pollutants can be prevented by blocking free radical processes in the obese lung with specific antioxidants that target MPO or MPO-derived oxidants, such as HOCl (Figure 2). Using genetically modified mice which are unable to release superoxide anions and superoxide-derived oxidants from NAD(P)H oxidases (NOX), it has been found that PM2.5 increases obesity and insulin resistance in vivo by NAD(P)H oxidase-derived superoxide anions [62]. Superoxide anions and their derived oxidants including hydrogen peroxide and peroxynitrite are known to suppress vascular endothelial function and lower insulin responsiveness [62]. The NAD(P)H oxidase, a membrane-bound enzyme complex, consists of multiple subunits including p22phox, p40phox, p47phox, p67phox, NoxO1, NoxA1, Rac1, and gp91phox-related unique isoforms of Nox [63]. The complex is normally latent in neutrophils and is activated to assemble in the membranes during respiratory burst [64]. The choice of Xu et. al. to target the p47phox subunit of NAD(P)H was based on its homology in both phagocytic and nonphagocytic inflammatory pathways that are implicated in NAD(P)H-mediated insulin resistance [65]. In an elegant series of experiments using C57BL6 and p47phox homozygous knockout mice, the investigators have systematically assembled evidence that PM2.5 exposure when the animals were 3-week-old young pups, induced a later life phenotype that manifested increase in abdominal fat, increased adipocyte size, and heightened inflammatory cellular response [65]. This study suggests that early life exposure to PM2.5 scale particulate pollution primes the system toward developing insulin resistance and a proinflammatory vascular phenotype in later life, likely via NAD(P)H oxidase–derived superoxide anions.
Therapeutic Targets
The link between adipose tissue inflammation, systemic inflammation and pulmonary neutrophilic inflammation should be interesting to break because pulmonary inflammation can contribute to the systemic pool of inflammation mediators that can worsen systemic inflammation, and consequently obesity-associated co-morbidities, such as insulin resistance (Figure 2). Obviously, the most desired therapeutic approach is that that can control appetite or increase energy expenditure avoiding the chronic positive energetic balance [66]. On another hand, change or transplant of microbiota may be a chance to revert changes in gut microflora in obese patients [67]. However, this option has been recently questioned after the US Food and Drug Administration informed the death of one patient by contamination with Clostridium difficile during a clinical trial. Consequently, more research is needed on this target.
Adipose tissue inflammation is a consequence of a metabolic inflammation process [10]. Macrophage activation by either FFAs or endotoxin should be targeted. The common link between these two stressors is the NF-kB signaling pathway [14], Table 1. Indeed, endotoxin and FFA (palmitic-, miristic- and stearic- fatty acids) activate innate immune cells by binding to TLR-4 and TLR-2, respectively, thus contribute to metabolic inflammation in the adipose tissue. Activation of TLRs in macrophages leads to signal transduction that induces NF-?B activation-the master regulator of the inflammatory response [14]. Tissue cell activation with production of chemokines may be anther target, because these attract monocytes that differentiate into M1 macrophages. MCP-1 and 4-1BB are interesting target and the use of peptidomimetics should be further explored to interfere in the binding to its receptors in monocytes [10.68,69] The microenvironment of metabolically irritated adipose tissue by FFAs may be another target. In the irritated and hypoxic environment of metabolically stressed adipose tissue, recruited monocytes differentiate into M1-inflammatory macrophages [12], which produce large quantities of pro-inflammatory cytokines and reactive biochemical species (reactive oxygen species, nitrogen reactive species, and macromolecule end-oxidation products) [10]. Thus, the first level of intervention should be improving the storage of FFAs [70,71], See Table 1.

Previously has been shown that Nrf2 is an important transcription factor controlling the expression of two transcription factors master regulators of adipogénesis [70]. These transcription factors are PPAR-? and CEBP-? [72]. These transcription factors control the expression of adipogenic enzymes that catalyze the synthesis of tryglicerides—the safest way to store excess FFAs [73]. Thus, by activating Nrf-2 circulating concentrations of FFAs can be reduced and systemic inflammation reduced as well as metabolic complications of obesity, such as pulmonary inflammation [71]. In this regard we have found that the nitrone spin trap DMPO can block alternative activation of macrophages by the endotoxin lipopolysaccharide [74-76]. This spin trap also causes transcriptomic changes related to an inhibition of the expression of genes related to inflammatory activation [74]. The inhibitory effect of DMPO on LPS-induced macrophage activation was due to the direct binding of the spin trap to four specific residues in the TIR domain of TLRs (TLR2) named BB-loop [75].
Myeloperoxidase and HOCl may be another target for preventing pulmonary neutrophilic inflammation in obesity (Table 1). Indeed, neutrophils, MPO and markers of HOCl-promoted oxidations have been observed in the lung of obese patients and in mouse models of diet-induced obesity and metabolic syndrome. Thus, the development of inhibitors of MPO or scavengers of HOCl may prove to be effective in preventing adipose tissue dysfunction (adipokine dysbalance, insulin resistance and cell death). Targeting the lung by using MPO inhibitors or HOCl scavengers may prove to be effective to prevent the contribution of pulmonary neutrophilic inflammation towards systemic inflammation, the leading cause of obesity-associated metabolic abnormalities (Figure 2). Thus, targeting inflammation and oxidative stress in the growing adipose tissue and lung is an attractive strategy to prevent obesity-associated co-morbidities and probably obesity associated cancers.
Concluding Remarks
Obesity is state of chronic low-grade inflammation caused by a chronic positive energy balance. This causes adipose tissue inflammation with infiltration of monocytes that acquire an inflammatory phenotype. The adipose tissue is inflamed and oxidants overwhelm the antioxidant capacity causing oxidative stress. Oxidized products as well as pro-inflammatory cytokines are secreted into the systemic circulation where it can activate neutrophils as well as activate NF-?B, and other inflammatory transcription factors leading to expression of neutrophil adhesion molecules in the lung microvasculature. Oxidative stress and inflammation in the lung increased the systemic pool of pro-inflammatory and end-oxidation products that reduce insulin sensitivity and cause other metabolic abnormalities-associated to obesity. Therapeutic strategies to reduce the pulmonary contribution towards systemic inflammation are needed. These should be focused in reducing neutrophilic inflammation in the lung microvasculature (see Table 1). They include improving fatty acid storage in adipose tissue by inducing Nrf-2, reducing inflammatory activation of adipose tissue macrophages, inhibiting MPO activity and scavenging HOCl in the lung. These approaches are active fields of research in our laboratories.
Acknowledgements
The informations reviewed herein was obtained with the following grants: Universidad nacional de San Luis, Argentina (PROICOs 100218 and 023418); Agencia Nacional para la Promoción de la Ciencia y la Tecnología, FONCYT (PICT 3369) and Consejo Nacional de Investigaciones Científicas y Técnicas (PIP916 y PUE229-201801-00013CO).
References
1. Saltiel AR, Olefsky JM. 2017. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. 127: 1-4. [Ref.:]
2. Karczewski J, Sledzinska E, Baturo A, et al. 2018. Obesity and inflammation. Eur Cytokine Netw. 29: 83-94
3. Boden G: 2008. Obesity and free fatty acids. Endocrinol Metab Clin North Am. 37: 635-646. [Ref.:]
4. Osborn O, Sears DD, Olefsky JM: Fat-induced inflammation unchecked. Cell Metab 2010;12: 553-554. [Ref.:]
5. Koliaki C, Liatis S, Kokkinos A. 2019. Obesity and cardiovascular disease: revisiting an old relationship. Metabolism. 92: 98-107. [Ref.:]
6. Nijhuis J, Rensen SS, Slaats Y, et al. 2009. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring). 17: 2014-2018. [Ref.:]
7. Hotamisligil GS. 2006. Inflammation and metabolic disorders. Nature. 444: 860-867. [Ref.:]
8. Hotamisligil GS, Shargill NS, Spiegelman BM. 1993. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 259: 87-91. [Ref.:]
9. Hersoug LG, Moller P, Loft S. 2018. Role of microbiota-derived lipopolysaccharide in adipose tissue inflammation, adipocyte size and pyroptosis during obesity. Nutr Res Rev. 31: 153-163. [Ref.:]
10. Lee YS, Wollam J, Olefsky JM. 2018. An Integrated View of Immunometabolism. Cell. 172: 22-40. [Ref.:]
11. Minamino T, Orimo M, Shimizu I, et al. 2009. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 15: 1082-1087. [Ref.:]
12. Lee CH, Lam KS. 2019. Obesity-induced insulin resistance and macrophage infiltration of the adipose tissue: A vicious cycle. J Diabetes Investig. 10: 29-31. [Ref.:]
13. Oh DY, Morinaga H, Talukdar S, et al. 2012. Increased macrophage migration into adipose tissue in obese mice. Diabetes. 61: 346-354. [Ref.:]
14. Solinas G, Karin M. 2010. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J. 24: 2596-2611. [Ref.:]
15. Trayhurn P. 2013. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev. 93: 1-21. [Ref.:]
16. Longo M, Zatterale F, Naderi J, et al. 2019. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int J Mol Sci. [Ref.:]
17. Perrotta F, Nigro E, Mollica M, et al. 2019. Pulmonary Hypertension and Obesity: Focus on Adiponectin. Int J Mol Sci. [Ref.:]
18. Janochova K, Haluzik M, Buzga M. 2019. Visceral fat and insulin resistance - what we know? Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 163: 19-27. [Ref.:]
19. Boucher J, Castan-Laurell I, Daviaud D, et al. 2005. Adipokine expression profile in adipocytes of different mouse models of obesity. Horm Metab Res. 37: 761-767. [Ref.:]
20. Ghadge AA, Khaire AA. 2019. Leptin as a predictive marker for metabolic syndrome. Cytokine. [Ref.:]
21. Zhao Q, Liu Y, Tan L, et al. 2018. Adiponectin administration alleviates DSS-induced colonic inflammation in Caco-2 cells and mice. Inflamm Res. 67: 663-670. [Ref.:]
22. Panagopoulou P, Galli-Tsinopoulou A, Fleva A, et al. 2008. Adiponectin and insulin resistance in childhood obesity. J Pediatr Gastroenterol Nutr. 47: 356-362. [Ref.:]
23. Kadowaki T, Yamauchi T, Kubota N, et al. 2007. Adiponectin and adiponectin receptors in obesity-linked insulin resistance. Novartis Found Symp. 286: 164-176. [Ref.:]
24. Ouchi N, Walsh K. 2007. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 380: 24-30. [Ref.:]
25. Akhter N, Madhoun A, Arefanian H, et al. 2019. Oxidative Stress Induces Expression of the Toll-Like Receptors (TLRs) 2 and 4 in the Human Peripheral Blood Mononuclear Cells: Implications for Metabolic Inflammation. Cell Physiol Biochem. 53: 1-18. [Ref.:]
26. Friesen M, Cowan CA. 2019. Adipocyte Metabolism and Insulin Signaling Perturbations: Insights from Genetics. Trends Endocrinol Metab. 30: 396-406. [Ref.:]
27. Kobayashi Y. 2006. Neutrophil infiltration and chemokines. Crit Rev Immunol. 26: 307-316. [Ref.:]
28. Shore SA, Lang JE, Kasahara DI, et al. 2009. Pulmonary responses to subacute ozone exposure in obese vs. lean mice. J Appl Physiol. 107: 1445-1452. [Ref.:]
29. Mafort TT, Rufino R, Costa CH, et al. 2016. Obesity: systemic and pulmonary complications, biochemical abnormalities, and impairment of lung function. Multidiscip Respir Med. 11:28. [Ref.:]
30. Koenig SM. 2001. Pulmonary complications of obesity. Am J Med Sci. 321: 249-279. [Ref.:]
31. Haque AK, Gadre S, Taylor J, et al. 2008. Pulmonary and cardiovascular complications of obesity: an autopsy study of 76 obese subjects. Arch Pathol Lab Med. 132: 1397-1404. [Ref.:]
32. Shore SA. 2010. Obesity, airway hyperresponsiveness, and inflammation. J Appl Physiol 108: 735-743. [Ref.:]
33. Nathan C. 2006. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 6: 173-182. [Ref.:]
34. Kolaczkowska E, Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 13: 159-175. [Ref.:]
35. Barnard JW, Biro MG, Lo SK, et al. 1995. Neutrophil inhibitory factor prevents neutrophil-dependent lung injury. J Immunol. 155: 4876-4881. [Ref.:]
36. Wang J. 2018. Neutrophils in tissue injury and repair. Cell Tissue Res. 371: 531-539. [Ref.:]
37. Herishanu Y, Rogowski O, Polliack A, et al. 2006. Leukocytosis in obese individuals: possible link in patients with unexplained persistent neutrophilia. Eur J Haematol. 76: 516-520. [Ref.:]
38. Mitchell LA, Gao J, Wal RV, et al. 2007. Pulmonary and systemic immune response to inhaled multiwalled carbon nanotubes. Toxicol Sci. 100: 203-214. [Ref.:]
39. Siela D. 2009. Pulmonary aspects of obesity in critical care. Crit Care Nurs Clin North Am. 21: 301-310. [Ref.:]
40. Christou KA, Christou GA, Karamoutsios A, et al. 2019. Metabolically Healthy Obesity Is Characterized by a Proinflammatory Phenotype of Circulating Monocyte Subsets. Metab Syndr Relat Disord. 17: 259-265. [Ref.:]
41. Reid PT, Donnelly SC, MacGregor IR, et al. 2000. Pulmonary endothelial permeability and circulating neutrophil-endothelial markers in patients undergoing oesophagogastrectomy. Crit Care Med. 28: 3161-3165. [Ref.:]
42. Carlos TM, Harlan JM. 1994. Leukocyte-endothelial adhesion molecules. Blood. 84: 2068-2101. [Ref.:]
43. Kahn BB. 2019. Adipose Tissue, Inter-Organ Communication, and the Path to Type 2 Diabetes: The 2016 Banting Medal for Scientific Achievement Lecture. Diabetes. 68: 3-14. [Ref.:]
44. Wagner E, Lim L, Mitzner W. 2003. Leukocyte transit through the airway circulation. Arch Physiol Biochem. 111: 345-346. [Ref.:]
45. Gee MH, Albertine KH. 1993. Neutrophil-endothelial cell interactions in the lung. Annu Rev Physiol. 55: 227-248. [Ref.:]
46. Wagner JG, Roth RA. 2000. Neutrophil migration mechanisms, with an emphasis on the pulmonary vasculature. Pharmacol Rev. 52: 349-374. [Ref.:]
47. Perfetto F, Mancuso F, Tarquini R. 2002. Leukocytosis and hyperleptinemia in obesity: is there a link? Haematologica. [Ref.:]
48. Haynes AP, Fletcher J. 1990. Neutrophil function tests. Baillieres Clin Haematol. 3: 871-887. [Ref.:]
49. Itoh H, Morihana Y, Cheong WK, et al. 1981. Neutrophilic polymorphonuclear leucocytes in obese children. Studies by Sudan Black B and electron microscope. Acta Pathol Jpn. 31: 167-177. [Ref.:]
50. Della Vedova MC, Soler Garcia FM, Muñoz MD, Fornes MW, Gomez Mejiba SE, Gomez NN, Ramirez DC: Diet-induced pulmonary inflammation and incipient fibrosis in mice: a possible role of neutrophilic inflammation. Inflammation 2019;In press: doi: 10.1007/s10753-10019-01051-10759
51. Gomez Garcia A, Rivera Rodriguez M, Gomez Alonso C, et al. 2015. Myeloperoxidase is associated with insulin resistance and inflammation in overweight subjects with first-degree relatives with type 2 diabetes mellitus. Diabetes Metab J. 39: 59-65. [Ref.:]
52. Nicholls SJ, Hazen SL. 2005. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 25: 1102-1111. [Ref.:]
53. Klebanoff SJ. 2005. Myeloperoxidase: friend and foe. J Leukoc Biol. 77: 598-625. [Ref.:]
54. Davies MJ, Hawkins CL, Pattison DI, et al. 2008. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid Redox Signal. 10: 1199-1234. [Ref.:]
55. Kettle AJ, Obinger C. 2000. Mammalian peroxidases brought into focus. Redox Rep. 5: 167-168. [Ref.:]
56. Kettle AJ, Albrett AM, Chapman AL, et al. 2014. Measuring chlorine bleach in biology and medicine. Biochim Biophys Acta. 1840:781-793. [Ref.:]
57. Gaut JP, Byun J, Tran HD, et al. 2002. Myeloperoxidase produces nitrating oxidants in vivo. J Clin Invest. 109: 1311-1319. [Ref.:]
58. Muzumdar H, Rao M. 2006. Pulmonary dysfunction and sleep apnea in morbid obesity. Pediatr Endocrinol Rev. 4:579-583. [Ref.:]
59. Nemmar A, Al-Maskari S, Ali BH, et al. 2007. Cardiovascular and lung inflammatory effects induced by systemically administered diesel exhaust particles in rats. Am J Physiol Lung Cell Mol Physiol. 292: 664-670. [Ref.:]
60. Pope CA, Burnett RT, Thurston GD, et al. 2004. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation. 109: 71-77. [Ref.:]
61. Shore SA. 2011. Environmental perturbations: obesity. Compr Physiol. 1: 263-282.
62. Zou MH. 2010. Is NAD(P)H oxidase a missing link for air pollution-enhanced obesity? Arterioscler Thromb Vasc Biol. 30: 2323-2324. [Ref.:]
63. Lambeth JD. 2004. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 4:181-189. [Ref.:]
64. Babior BM, Lambeth JD, Nauseef W. 2002. The neutrophil NADPH oxidase. Arch Biochem Biophys. 397: 342-344. [Ref.:]
65. Xu H, Barnes GT, Yang Q, et al. 2003. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 112: 1821-1830. [Ref.:]
66. Chhabra KH, Adams JM, Jones GL, et al. 2016. Reprogramming the body weight set point by a reciprocal interaction of hypothalamic leptin sensitivity and Pomc gene expression reverts extreme obesity. Mol Metab. 5: 869-881. [Ref.:]
67. Kulecka M, Paziewska A, Zeber-Lubecka N, et al. 2016. Prolonged transfer of feces from the lean mice modulates gut microbiota in obese mice. Nutr Metab (Lond). 13:57. [Ref.:]
68. Kim CS, Kim JG, Lee BJ, et al. 2011. Deficiency for Costimulatory Receptor 4-1BB Protects Against Obesity-Induced Inflammation and Metabolic Disorders. Diabetes. 60: 3159-3168. [Ref.:]
69. Gomez-Mejiba SE, Ramirez DC. 2012. Comment on: Kim et al. Deficiency for costimulatory receptor 4-1BB protects against obesity-induced inflammation and metabolic disorders. Diabetes. 60: 3159-3168. [Ref.:]
70. Pi J, Leung L, Xue P, et al. 2010. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J Biol Chem. 285: 9292-9300. [Ref.:]
71. Aleksunes LM, Reisman SA, Yeager RL, et al. 2010. Nuclear factor erythroid 2-related factor 2 deletion impairs glucose tolerance and exacerbates hyperglycemia in type 1 diabetic mice. J Pharmacol Exp Ther. 333: 140-151. [Ref.:]
72. Gauvreau D, Gupta A, Fisette A, et al. 2013. Deficiency of C5L2 Increases Macrophage Infiltration and Alters Adipose Tissue Function in Mice. PLoS One. [Ref.:]
73. Kang K, Hatano B, Lee CH. 2007. PPAR delta agonists and metabolic diseases. Curr Atheroscler Rep. 9: 72-77. [Ref.:]
74. Munoz MD, Della Vedova MC, Bushel PR, et al. 2018. The nitrone spin trap 5,5-dimethyl-1-pyrroline N-oxide dampens lipopolysaccharide-induced transcriptomic changes in macrophages. Inflamm Res. 67: 515-530. [Ref.:]
75. Munoz MD, Gutierrez LJ, Delignat S, et al. 2019. The nitrone spin trap 5,5dimethyl1pyrroline Noxide binds to toll-like receptor-2-TIR-BB-loop domain and dampens downstream inflammatory signaling. Biochim Biophys Acta Mol Basis Dis. 1865:1152-1159. [Ref.:]
76. Zhai Z, Gomez-Mejiba SE, Zhu H, et al. 2012. The spin trap 5,5-dimethyl-1-pyrroline N-oxide inhibits lipopolysaccharide-induced inflammatory response in RAW 264.7 cells. Life Sci 90: 432-439. [Ref.:]


















